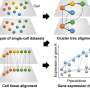
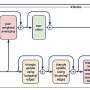
UMAP of cells in MERFISH—fetal liver data. (b) Imputation of MERFISH data with scRNA-seq increases the number of genes that can be modeled with NCEMs, including receptor genes, ligand genes, and ligand–receptor pairs. (c) Distribution of selected marker genes that are both observed in scRNA-seq and in MERFISH over cell types. Credit: Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01467-z")
Modeling ligand–receptor signaling with NCEM. (a) UMAP of cells in MERFISH—fetal liver data. (b) Imputation of MERFISH data with scRNA-seq increases the number of genes that can be modeled with NCEMs, including receptor genes, ligand genes, and ligand–receptor pairs. (c) Distribution of selected marker genes that are both observed in scRNA-seq and in MERFISH over cell types. Credit: Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01467-z
Cells interact in various different ways and on multiple length-scales. The interaction of a cell with its tissue niche can be described through cell communication events. To understand these events, researchers around the world create models, based on different strategies. The knowledge is crucial to understand and identify emerging phenomena in tissue microenvironments, such as genetic changes in a tumor.
The issue is many of the models are based on dissociated cells, meaning that the cells are separated to individual cells when being analyzed and are no longer integrated in their natural environment. Other models are limited to receptor-ligand signaling, a certain type of communication between cells.
These models therefore ignore the spatial proximity of a group of cells (a niche) in their natural tissue environment. Researchers led by Fabian Theis from the Computational Health Center at Helmholtz Munich and Technical University of Munich (TUM) have now developed a new method, that defines the complexity and improves the understanding of cell communication: The node-centric expression models (NCEM).
A flexible framework
NCEM is a computational method based on graph neural networks, which combine transcriptomic variance attribution and cell communication modeling in a single model of tissue niches.
The model is therefore able to predict a cell's gene expression profile based on the presence of surrounding cell types. In addition, it estimates the effect of a tissue niche composition on gene expression in an unbiased manner from spatial molecular profiling data.
In their model, the researchers developed a flexible framework to explain gene expression variations observable in spatial transcriptomics, a technology providing spatially-resolved gene expression information. Gene expression variations can then be associated to known molecular processes associated with cell communication events.
They showed that NCEMs robustly identify cell-cell dependencies across different spatial transcriptomics technologies and at length scales that are characteristic for known communication mechanisms. With this method, first authors David Fischer and Anna Schaar were thereby able to recover signatures of molecular processes, that are known to underlie cell communication.
A novel way to identify cell communication
The framework constraints communication events to cells that are proximal in space. The identified dependencies are not limited to ligand-receptor-based communication but can also explain, for example, physical interactions or metabolite exchange.
NCEM is a flexible computational method that can be extended to more complex data sets, as for example 3D spatial transcriptomics data and higher-throughput data. It therefore provides a flexible toolset for the analysis of cell-cell-communication in space. The novel methodology complements recent efforts on characterizing gene expression in individual cells in single-cell "atlas" projects by accounting in this particular case for the tissue niche.
The research is published in Nature Biotechnology.
More information: Fabian Theis, Modeling intercellular communication in tissues using spatial graphs of cells, Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01467-z. www.nature.com/articles/s41587-022-01467-z
Journal information: Nature Biotechnology
Provided by Helmholtz Association of German Research Centres