Computational approach enables spatial mapping of single-cell data within tissues
 and the ribose-phosphate backbone (blue). Note that this is a single strand of RNA that folds back upon itself. Credit: Vossman/ Wikipedia")
A new computational approach developed by researchers at The University of Texas MD Anderson Cancer Center successfully combines data from parallel gene-expression profiling methods to create spatial maps of a given tissue at single-cell resolution. The resulting maps can provide unique biological insights into the cancer microenvironment and many other tissue types.
The study was published today in Nature Biotechnology and will be presented at the upcoming American Association for Cancer Research (AACR) Annual Meeting 2022 (Abstract 2129).
The tool, called CellTrek, uses data from single-cell RNA sequencing (scRNA-seq) together with that of spatial transcriptomics (ST) assays—which measure spatial gene expression in many small groups of cells—to accurately pinpoint the location of individual cell types within a tissue. The researchers presented findings from analysis of kidney and brain tissues as well as samples of ductal carcincoma in situ (DCIS) breast cancer.
"Single-cell RNA sequencing provides tremendous information about the cells within a tissue, but, ultimately, you want to know where these cells are distributed, particularly in tumor samples," said senior author Nicholas Navin, Ph.D., professor of Genetics and Bioinformatics & Computational Biology. "This tool allows us to answer that question with an unbiased approach that improves upon currently available spatial mapping techniques."
Single-cell RNA sequencing is an established method to analyze the gene expression of many individual cells from a sample, but it cannot provide information on the location of cells within a tissue. On the other hand, ST assays can measure spatial gene expression by analyzing many small groups of cells across a tissue but are not capable of providing single-cell resolution.
Current computational approaches, known as deconvolution techniques, can identify different cell types present from ST data, but they are not capable of providing detailed information at the single-cell level, Navin explained.
Therefore, co-first authors Runmin Wei, Ph.D., and Siyuan He of the Navin Laboratory led the efforts to develop CellTrek as a tool to combine the unique advantages of scRNA-seq and ST assays and create accurate spatial maps of tissue samples.
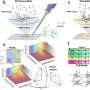
Using publicly available scRNA-seq and ST data from brain and kidney tissues, the researchers demonstrated that CellTrek achieved the most accurate and detailed spatial resolution of the methods evaluated. The CellTrek approach also was able to distinguish subtle gene expression differences within the same cell type to gain information on their heterogeneity within a sample.
The researchers also collaborated with Savitri Krishnamurthy, M.D., professor of Pathology, to apply CellTrek to study DCIS breast cancer tissues. In an analysis of 6,800 single cells and 1,500 ST regions from a single DCIS sample, the team learned that different subgroups of tumor cells were evolving in unique patterns within specific regions of the tumor. Analysis of a second DCIS sample demonstrated the ability of CellTrek to reconstruct the spatial tumor-immune microenvironment within a tumor tissue.
"While this approach is not restricted to analyzing tumor tissues, there are obvious applications for better understanding cancer," Navin said. "Pathology really drives cancer diagnoses and, with this tool, we're able to map molecular data on top of pathological data to allow even deeper classifications of tumors and to better guide treatment approaches."
Collaborating MD Anderson authors include Shanshan Bai, Emi Sei, Ph.D., and Min Hu, all of Genetics; and Ken Chen, Ph.D., of Bioinformatics. Additional authors include Alastair Thompson, M.D., of Baylor College of Medicine, Houston. The authors have no conflicts of interest.
More information: Nicholas Navin, Spatial charting of single-cell transcriptomes in tissues, Nature Biotechnology (2022). DOI: 10.1038/s41587-022-01233-1. www.nature.com/articles/s41587-022-01233-1
Journal information: Nature Biotechnology
Provided by University of Texas M. D. Anderson Cancer Center