Neighboring cell types influence single-cell gene expression variability

Sometimes you don't know what you're looking for until you find it; and this is especially true when it comes to the huge datasets that can be generated using modern sequencing techniques. Now, researchers from Japan report the development of a statistical framework that can perform unbiased extraction of biologically relevant cell-cell communications from a sea of spatial gene expression data.
In a study published in September in Bioinformatics, researchers from the University of Tsukuba have revealed that a new statistical analysis method can accurately identify cell-cell communication affecting gene expression at the single-cell level.
Communication between cells regulates gene expression in ways that are crucial for normal function as well as disease development. While single-cell RNA sequencing and spatially resolved transcriptomics can provide some insight into this communication, current methods for analyzing these types of data have some important limitations.
"Most existing statistical analysis methods do not account for the spatial organization of cells within an organ composed of various cell types," states Associate Professor Haruka Ozaki, senior author of the study. "However, the location of cells, the number of cells, and the cell types in the vicinity affect gene expression in neighboring cells."
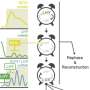
To capture this complexity, the researchers created a statistical framework called CCPLS (Cell-Cell communications analysis by Partial Least Square regression modeling) that analyzes spatial gene expression data at single-cell resolution. The aim of this system was to identify and quantify the influence of neighboring cell types on cell-to-cell variability in gene expression.
"We first applied CCPLS to a simulated data set and found that it accurately estimated the effects of multiple neighboring cell types on gene expression," says Associate Professor Ozaki. "Then we applied the system to a real-world data set and showed that astrocytes promote differentiation of oligodendrocyte precursor cells into oligodendrocytes, which is consistent with earlier mouse experiments."
Then, CCPLS was applied to another real-world data set containing gene expression data from nine different cell types found in the colon. The analysis showed that epithelial cell development of immature B cells occurs through communication with IgA B cells, which has not been previously reported.
"Our findings show that CCPLS can be used to extract biologically relevant insights about cell-cell communications from complex data sets," states Professor Ozaki.
Given that CCPLS outperformed an existing statistical framework in identifying gene expression variability regulated by cell-cell communication, it is likely that it will be a highly useful tool for data set analysis in the future. It may be particularly effective for exploring drug targets and in cases where cell arrangement causes changes to gene expression.
More information: Takaho Tsuchiya et al, CCPLS reveals cell-type-specific spatial dependence of transcriptomes in single cells, Bioinformatics (2022). DOI: 10.1093/bioinformatics/btac599
Journal information: Bioinformatics
Provided by University of Tsukuba