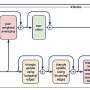
Preprocessing module that consists of finding the binding sites of proteins; (2) AttentionSiteDTI deep learning module, where we construct graph representations of ligands' SMILE and proteins' binding sites, and we create a graph convolutional neural network armed with an attention pooling mechanism to extract learnable embeddings from graphs, as well as a self-attention mechanism to learn relationship between ligands and proteins' binding sites; (3) Prediction module to predict unknown interaction in a drug–target pair, which can address both classification and regression tasks; (4) Interpretation module to provide a deeper understanding of which binding sites of a target protein are more probable to bind with a given ligand. (5) In-lab validations, where we compare our computationally predicted results with experimentally observed (measured) drug–target interactions in the laboratory to test and validate the practical potential of our proposed model. Credit: Briefings in Bioinformatics (2022). DOI: 10.1093/bib/bbac272")
Our proposed framework includes five main modules: (1) Preprocessing module that consists of finding the binding sites of proteins; (2) AttentionSiteDTI deep learning module, where we construct graph representations of ligands' SMILE and proteins' binding sites, and we create a graph convolutional neural network armed with an attention pooling mechanism to extract learnable embeddings from graphs, as well as a self-attention mechanism to learn relationship between ligands and proteins' binding sites; (3) Prediction module to predict unknown interaction in a drug–target pair, which can address both classification and regression tasks; (4) Interpretation module to provide a deeper understanding of which binding sites of a target protein are more probable to bind with a given ligand. (5) In-lab validations, where we compare our computationally predicted results with experimentally observed (measured) drug–target interactions in the laboratory to test and validate the practical potential of our proposed model. Credit: Briefings in Bioinformatics (2022). DOI: 10.1093/bib/bbac272
Developing life-saving medicines can take billions of dollars and decades of time, but University of Central Florida researchers are aiming to speed up this process with a new artificial intelligence-based drug screening process they've developed.
Using a method that models drug and target protein interactions using natural language processing techniques, the researchers achieved up to 97% accuracy in identifying promising drug candidates. The results were published recently in the journal Briefings in Bioinformatics.
The technique represents drug–protein interactions through words for each protein binding site and uses deep learning to extract the features that govern the complex interactions between the two.
"With AI becoming more available, this has become something that AI can tackle," says study co-author Ozlem Garibay, an assistant professor in UCF's Department of Industrial Engineering and Management Systems. "You can try out so many variations of proteins and drug interactions and find out which are more likely to bind or not."
The model they've developed, known as AttentionSiteDTI, is the first to be interpretable using the language of protein binding sites.
The work is important because it will help drug designers identify critical protein binding sites along with their functional properties, which is key to determining if a drug will be effective.
The researchers made the achievement by devising a self-attention mechanism that makes the model learn which parts of the protein interact with the drug compounds, while achieving state-of-the-art prediction performance.
The mechanism's self-attention ability works by selectively focusing on the most relevant parts of the protein.
The researchers validated their model using in-lab experiments that measured binding interactions between compounds and proteins and then compared the results with the ones their model computationally predicted. As drugs to treat COVID are still of interest, the experiments also included testing and validating drug compounds that would bind to a spike protein of the SARS-CoV2 virus.
Garibay says the high agreement between the lab results and the computational predictions illustrates the potential of AttentionSiteDTI to pre-screen potentially effective drug compounds and accelerate the exploration of new medicines and the repurposing of existing ones.
"This high impact research was only possible due to interdisciplinary collaboration between materials engineering and AI/ML and Computer Scientists to address COVID related discovery," says Sudipta Seal, study co-author and chair of UCF's Department of Materials Science and Engineering.
Mehdi Yazdani-Jahromi, a doctoral student in UCF's College of Engineering and Computer Science and the study's lead author, says the work is introducing a new direction in drug pre-screening.
"This enables researchers to use AI to identify drugs more accurately to respond quickly to new diseases," Yazdani-Jahromi says. "This method also allows the researchers to identify the best binding site of a virus's protein to focus on in drug design."
"The next step of our research is going to be designing novel drugs using the power of AI," he says. "This naturally can be the next step to be prepared for a pandemic."
More information: Mehdi Yazdani-Jahromi et al, AttentionSiteDTI: an interpretable graph-based model for drug-target interaction prediction using NLP sentence-level relation classification, Briefings in Bioinformatics (2022). DOI: 10.1093/bib/bbac272
Provided by University of Central Florida