. Credit: Laboratory of Genome Maintenance at The Rockefeller University/Molecular Cell")
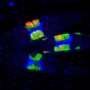
To confirm that a defect in RAD51 interfered with cells' ability to fix misplaced links between DNA strands, researchers treated patient cells with an agent to cause such links to form. The cells failed to repair them, producing broken chromosomes that fused with one another (red arrows). Credit: Laboratory of Genome Maintenance at The Rockefeller University/Molecular Cell
Dividing cells are prone to errors, and so they must be prepared to summon sophisticated emergency systems to deal with potential damage. One type of division-derailing mishap can occur when assault by certain chemicals causes two strands of DNA to permanently connect when they shouldn't, in what scientists call interstrand crosslinks (ICLs). Properly fixing these crosslinks is crucial to preventing cancer, maintaining tissues, and fertility.
To better understand how a cell finds and fixes these misplaced crosslinks, researchers at The Rockefeller University and their colleagues are examining the genomes of patients in whom the repair process is defective. In two separate studies, the most recent described in Molecular Cell on August 6, they have identified two new genes in which mutations can produce one such rare genetic disorder, Fanconi anemia, and so revealed new insights on this critical repair pathway.
"Our work began, as it often does, with samples and histories from patients. In these cases, we had two patients who each represented a sort of mystery: They had symptoms of Fanconi anemia, but no genetic cause yet identified," says senior author Agata Smogorzewska, associate professor and head of the Laboratory of Genome Maintenance. "Our investigation led us to discover a defective RAD51 protein in one patient, and a similarly dysfunctional protein UBE2T in the other."
The genes that code for RAD51 and UBE2T—along with many other genes linked to Fanconi anemia in previous studies—contribute to a repair process known as interstrand crosslink repair, which fixes a misplaced attachment between two strands of DNA. Caused by chemical agents, including often used chemotherapies like cisplatin; chemicals called aldehydes that occur naturally within cells, and nitrous acid formed after eating nitrates, ICLs block the replication of DNA, making it impossible for cells to accurately copy their genomes as they divide. The ICL repair process is very sophisticated and uses multiple enzymes that cut away the connection between the DNA strands, freeing them up and allowing the cells to grow.
The genome is at constant risk of forming ICLs, and defects in the ICL repair pathway can produce a constellation of symptoms associated with Fanconi anemia: a predisposition to cancer, failure of the stem cells in bone marrow responsible for producing blood cells, infertility, as well as developmental defects.
In the RAD51 research, supported by the Starr Cancer Consortium, first author Anderson Wang, a postdoctoral fellow in the Smogorzewska laboratory and his colleagues set out to determine the cause of the Fanconi anemia-like symptoms of a girl in the university's International Fanconi Anemia Registry. When they sequenced the protein-coding genes in her genome, they found mutations in one of two copies of the gene for the protein RAD51—a surprising culprit. This protein was already known to be important for another DNA repair process, called homologous recombination, in which a missing section of DNA is replaced using its sister strand as a template. Homologous recombination is thought to be used during the last step of ICL repair, after the crosslink has been cut.
But because only one copy of the RAD51 gene was partially defective, her cells could still perform homologous recombination, but not ICL repair. If both copies of RAD51, which is essential for life, had been defective, the girl would never have been born.
To show that the defective copy of the RAD51 gene was indeed responsible for her symptoms, the researchers genetically engineered the patient's own cells to remove the defect, which restored their ability to fix ICLs. Further experiments on the patient's cells —including biochemical work conducted by coauthor Stephen Kowalczykowski's lab at the University of California, Davis—lead the researchers to suspect that RAD51 plays a role outside of homologous recombination, by tamping down the activity of two enzymes that degrade the DNA at the ICL. When RAD51 is defective, these enzymes (DNA2 and WRN) become overly destructive.
In the UBE2T study, published July 7 in in Cell Reports, the team, including first author Kimberly Rickman, a biomedical fellow in Smogorzewska's lab, found that mutations in a gene for a protein named UBE2T explained the Fanconi anemia symptoms seen in another registry patient. While it was already known that UBE2T is involved in activating ICL repair, the discovery that these mutations could produce Fanconi anemia revealed the protein is an irreplaceable player in the pathway.
"Although we have discovered new causes for this devastating but very rare genetic disease, the implications of this work go much further. By identifying new disruptions to this repair pathway, we can better understand the mechanisms of an event that is crucial to every cell division—a process that occurs constantly within the human body throughout a lifetime," Smogorzewska says.
Journal information: Cell Reports , Molecular Cell
Provided by Rockefeller University