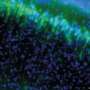
Luminescence and fluorescence images of subcellular structures: nuclei in interphase (CNL-H2B and ONL-H2B), F-actin [inositol trisphosphate 3-kinase A (ITPKA) -ONL], microtubules (β-tubulin-ONL), peroxisomes [peroxisome targeting signal 2 (PTS2) -CNL], and focal adhesions (zyxin-ONL). (B) Subnuclear structures visualized by dual-color luminescence imaging: CNL-fibrillarin (nucleoli) and ONL-H2B. Each luminescence signal was separated by linear unmixing. (Scale bars: 10 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.")
Luminescence imaging of intracellular microstructures. (A) Luminescence and fluorescence images of subcellular structures: nuclei in interphase (CNL-H2B and ONL-H2B), F-actin [inositol trisphosphate 3-kinase A (ITPKA) -ONL], microtubules (β-tubulin-ONL), peroxisomes [peroxisome targeting signal 2 (PTS2) -CNL], and focal adhesions (zyxin-ONL). (B) Subnuclear structures visualized by dual-color luminescence imaging: CNL-fibrillarin (nucleoli) and ONL-H2B. Each luminescence signal was separated by linear unmixing. (Scale bars: 10 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.
While fluorescence imaging (in which external light is used to excite a specimen that then emits light in response) is essential in cell biology, it has a number of significant drawbacks, including autofluorescence, phototoxicity and photobleaching, resulting from that excitation light. In addition, fluorescence imaging has the unfortunate side effect of triggering cellular activation when combined with optogenetics – an otherwise extremely valuable tool. On the other hand, luminescence (in this case, a type of chemiluminescence called bioluminescence) imaging doesn't require light activation, and so eschews these issues – but currently suffers from low brightness and poor color variants.
Recently, however, scientists at RIKEN and Osaka University extended their previous development of a bright yellowish-green luminescent protein Nano-lantern to devise bright cyan and orange luminescent proteins some 20 times brighter than previously possible with wild-type (i.e., naturally-occurring) Renilla luciferase, or Rluc – an oxidative enzyme associated with a luciferin-binding protein – found in a type of soft coral known as a sea pansy. (Luciferins are organic substances, found in luminescent organisms, which produce a near-heatless light upon oxidation.) Specifically, the researchers accomplished this by bioluminescence resonance energy transfer (BRET) from enhanced Renilla luciferase to a fluorescent protein, stating that their proof-of-principle experiments show that luminescence imaging has become a practical alternative when the side effects by the excitation light are not negligible – for example, when the samples are very sensitive to photodamage – and that the most effective future application of luminescence imaging lies in combining it with optogenetics, since the latter's external light illumination can be reserved for optical stimulation.
Dr. Yasushi Okada discussed the paper that he, Dr. Akira Takai, Dr. Takeharu Nagai, and their colleagues published in Proceedings of the National Academy of Sciences, starting with the challenges of developing cyan and orange luminescent proteins approximately 20 times brighter than wild-type Renilla luciferase. "Making the cyan version was straightforward," Okada tells Phys.org. "We acquired the best available cyan fluorescent protein, mTurquoise2," or mTq2, "from Dr Joachim Goedhart at University of Amsterdam. I was so impressed with the title of his paper1 that I immediately requested the plasmid – and he said that my request was the first he received. Several weeks after, we started working with our colleague Dr. Takeharu Nagai on yellow lanterns and soon came up with the idea of swapping the mVenus used in the yellow Nano-lantern," or YNL, "with mTq2 – and it worked excellently, producing the cyan Nano-lantern," or CNL.
That said, Okada adds that developing the orange Nano-lantern (ONL) took trial-and-error. "We initially planned to use a large Stokes shift orange fluorescent protein like LSSmOrange2, but it didn't work well." A Stokes shift, which is essential in fluorescence imaging, refers to the difference between the energy (i.e., wavelength) of the excitation and emitted photons – and a large Stokes shift typically indicates a greater difference and thereby easier detection. "We therefore tested all possible combinations of available orange to red fluorescent proteins and Rluc variants – hundreds of them – and we finally identified the best combination." That combination became the orange Nano-lantern.
The paper describes an application in which three gene expression reporters (genes attached to a regulatory sequence of a gene being investigated) were introduced into the genome of photosensitive embryonic stem (ES) cells by using transposase mediated insertion. (Transposase is an enzyme that binds to the end of a transposon – a DNA sequence that can change its position within the genome – and catalyzes transposon movement to and insertion in another part of the genome.) The triple positive cells were then screened by fluorescence-activated cell sorting (FACS), a method for sorting a heterogeneous mixture of biological cells into two or more containers, one cell at a time, based upon the specific light scattering and fluorescent characteristics of each cell. "Here, the dual use of our Nano-lanterns was very useful," Okada recounts. "They can be observed both by fluorescence and luminescence, so we can therefore use FACS for screening for the triple positive cells. "Our major imaging challenge was the consumption of the substrate, coelenterazine." The light detected in bioluminescence imaging is produced when luciferase is expressed in vivo and oxidizes a molecular substrate, while coelenterazine is the luciferin found in many aquatic organisms.
"Although we've developed diacetyl coelenterazine," in which protective acetyl groups inhibit autoxidation, allowing it to be added at higher concentrations without increasing background autoluminescence, "we needed to supply it continuously for long-term observation that exceed 12 hours. Our colleague and co-author Dr. Tomonobu Watanabe performed the imaging with his handmade perfusion system."
Another issue in luminescence imaging with Nano-lanterns is that its signal intensity is still over 100 times weaker than that of fluorescent proteins. "Yes, our Nano-lanterns are still darker than fluorescent proteins," Okada agrees, "but fluorescent protein brightness is proportional to the excitation power until saturation. With strong excitation close to saturation, a single fluorescent protein molecule can emit more than 107 photons per second, while our Nano-lantern emits less than 10 photons per second. However, to avoid photobleaching we normally do not use such strong excitation beams for fluorescent live cell imaging. Even for a single molecule live cell imaging" he points out, "we reduce the excitation power to get only 100-1000 photons per second from a single fluorescent protein. In short, the "more than 100 times weaker" comparison in the article is based on this calculation – and considering that most of live cell fluorescent imaging is normally done with much weaker excitation beams, Nano-lantern brightness in such applications will be much closer to that of fluorescent proteins."
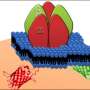
 Oct4 (CNL), (B) Nanog (YNL), and (C) Sox2 (ONL) were separated by linear unmixing and overlaid with the bright-field image. (D) Signals of CNL, YNL, and ONL were merged and overlaid with the bright-field image. (Scale bars: 100 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.")
Inhomogeneous expression of pluripotency markers in a single colony of ES cells. Luminescence signals of reporters for (A) Oct4 (CNL), (B) Nanog (YNL), and (C) Sox2 (ONL) were separated by linear unmixing and overlaid with the bright-field image. (D) Signals of CNL, YNL, and ONL were merged and overlaid with the bright-field image. (Scale bars: 100 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.
Okada acknowledges that since luminescence imaging requires a molecular substrate instead of excitation light, and the supply and consumption of the substrate limit imaging time, further increases in luminescence imaging light output would require higher enzymatic turnover rate because the number of photons per single enzymatic reaction does not exceed 1. The substrate would then be exhausted more rapidly, making substrate supply the more important challenge. The next frontier is therefore enhancing substrate synthesis or recycling. "For the bacterial luciferase and firefly luciferase" (the former is found in the Photobacterium species Vibrio fischeri, V. haweyi, and V. harveyi) "the supply systems for their substrate are already reconstituted in bacteria or plants. It won't take much time before the coelenterazine regeneration system will be reconstituted in vitro or in vivo."
Luminescence imaging also lacks optical sectioning capabilities. "If you use a conventional microscope," Okada explains, "the axial resolution of luminescence imaging is much poorer than fluorescence imaging. (Axial resolution refers to the ability to image at various depths in 3D imaging.) "However, we have several ideas to increase axial resolution, because it's critically important for applications like in vivo imaging."
In fluorescent imaging, different colors of fluorescent proteins can easily be separated by the combination of excitation and emission filters, whereas in luminescence imaging signals from different colors of Nano-lanterns can be separated only by the emission spectrum, and therefore require (albeit a simpler) linear unmixing algorithm. "While the emission spectra of luminescent proteins were much broader than fluorescent proteins, Nano-lantern emission is essentially same as that of the fluorescent proteins," Okada tells Phys.org. "As a result, the image is sharp enough for efficient and accurate separation by the linear unmixing algorithm. Furthermore, the background is completely dark, because luminescence imaging is free from autofluorescence, making the luminescence images are even more suitable for post-processing unmixing." (Autofluorescence is the natural emission of light by biological structures after they absorb light, and is used to distinguish the light originating from artificially added fluorescent markers called fluorophores.) "Related to this background issue," relates Okada, "we received a query from our paper's PNAS editor that the images were apparently manipulated because the background is homogeneously dark. We pointed out that this extremely low level background is a feature of luminescence imaging that makes it more sensitive and quantitative."
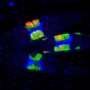
Continuous luminescence imaging of LAMP1-ONL in an MDCKII cell. Images with 2-s exposure were repeated for 7.8 min. Playback speed is 200-fold. (Scale bar: 10 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.
Despite the challenges they met and still face, Okada says that "From our experiences in fluorescence imaging, as well as the history of the development of fluorescent proteins, it was clear from the beginning that development of new color variants was absolutely required to extend the application of luminescence imaging – and fortunately, the production methodologies for color variants were evident from the BRET-based design of the yellow Nano-lantern."
It is also the case that Nano-lantern luminescence imaging has a range of significant benefits relative to current luminescence imaging, one being its ability to visualize rapid dynamics of endosomes and peroxisomes at a temporal resolution of one second. (An endosome is a membrane-bounded compartment inside eukaryotic cells involved in endocytic membrane transport, a pathway by which by which cells absorb molecules by engulfing them; a peroxisome is a small, membrane-enclosed organelle containing enzymes involved in a variety of metabolic reactions.) "Previous luminescence probes were very dim, and their imaging normally required extremely long exposure time – typically 10 minutes or longer," Okada points out. "Therefore, extremely low noise cameras, such as those cooled with liquid nitrogen, were required. Alternatively, you have to compromise spatial resolution to increase the frame rate. Therefore, it was impossible to observe small, but rapidly moving structures with luminescence. However, when expressed in animal cells our Nano-lantern is much brighter by several orders of magnitude." In fact, Okada relates, when his collaborators first used Nano-lanterns, they often reported that they could not take images – but this occurred because that their imaging system was optimized for conventional luminescence probes, and so was oversaturated by the Nano-lantern's brightness. "I often advised them to image the sample as if it were fluorescently stained, but without excitation light. That solved the problem."
The endosome and peroxisome experiments were in essence designed to demonstrate Nano-lantern brightness. Since both endosomes and peroxisomes are small intracellular structures with <1 micrometer diameters, with fluorescent staining they usually appear as diffraction limited spots, but actually are moving rapidly within the cell – typically at 1-10 micrometers per second. This means that both spatial and temporal resolutions are required to observe these organelles. "The experiment itself was quite easy," Okada says. "We made DNA constructs with endosome- or peroxisome- targeting signaling fused to the Nano-lantern and introduced them into the cell. Then, the cell expressed the probes, and endosomes or peroxisomes are labeled with the Nano-lantern." By so doing, the scientists obtained high-quality images with their first experiment. "Perhaps more importantly, it's strong proof that our Nano-lantern is bright enough to image subcellular structures at ~1 second time resolution."
Nano-lantern luminescence imaging also allows continuously imaging the slow dynamics of focal adhesions for more than several hours without photobleaching or photodamage. "One of the big technical challenges with fluorescence live imaging is photodamage resulting from its strong excitation light," Okada stresses. "The strong illumination first damages the fluorescent molecules themselves, which is called photobleaching, so the fluorescence signal decays during observation. Secondly, during the processes of photobleaching, radicals such as reactive oxygen are produced and can attack nearby molecules. Thirdly, the excitation light can be absorbed by molecules in the cell other than the fluorescent probes, and can thereby trigger other damaging processes." While the exact mechanisms of these types of photodamage are as yet unclear, the effects are obvious in some cell states or tissues. For example, in dividing cells cell division stops; migrating cells often stop moving; embryonic stem cells stop growing and lose pluripotency; and mammalian eggs or early embryos stop developing.
"This experiment demonstrated that luminescence imaging is free from these potential photodamage because it does not require excitation light," Okada details. "We chose moving cells, because they are known to stop movement by strong illumination. We labeled focal adhesions, the essential structures for the cell to attach to the substrate surface." (Focal adhesions are large macromolecular assemblies through which mechanical force and regulatory signals are transmitted between a cell and its extracellular matrix.) "They are also structures with ~1 micrometer size, and high spatial resolution is required to visualize them. After taking luminescence images continuously for six hours, the cells were healthy, moving directionally by extending new pseudopods. The dynamic turnover of focal adhesions was clearly observed." (In cells or some unicellular organisms, pseudopods are temporary cytoplasm projections that provide locomotion.) "Conventionally, such imaging requires time-lapse recording. The cells were illuminated <1 sec with an interval of 5-10 min, so as to minimize photodamage – but if you were to observe continuously with fluorescence, the cell stops moving within 10 minutes and dies."
Continuous luminescence imaging of ONL-PTS1 in an MDCKII cell. Images with 3-s exposure were repeated for 8.8 min. Playback speed is 200-fold. (Scale bar: 10 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.
Combining Nano-lanterns with optical manipulation tools – such as optogenetics, in which light-controllable channel proteins or light-sensing proteins are used to control cellular states – promises to be the most effective future application of luminescence imaging because the external light illumination can be reserved for optical stimulation. In the current research, blue to green light is used for the control, but the same color of light is used for fluorescent probe excitation – meaning that fluorescence imaging often conflicts with optogenetic tools. "For example," Okada illustrates, "it's been difficult to monitor Ca2+ calcium ion responses after optical stimulation of the channel protein because the latter is controlled with blue light while the fluorescent Ca2+ probe requires blue light for excitation." (Calcium imaging allows, for example, studying neuronal activity in hundreds of neurons and glial cells within neuronal circuits.) "Our Nano-lantern can solve this conflict: By using a Nano-lantern-based Ca2+ probe, one can observe the Ca2+ response without any external excitation light – so the external illumination can be reserved for the optical stimulation." Okada adds that the Ca2+ indicators that are fully compatible with optogenetic tools and other optical manipulations were developed using the cyan Nano-lantern and orange Nano-lantern by Dr. Masahiro Nakano in Dr. Nagai's lab.
Currently, Okada summarizes, fluorescent probes are mainly used in three key areas:
1. localization and dynamics of specific target proteins or structures
2. monitoring gene expression
3. indicators for physical or chemical states, such as intracellular Ca2+ concentration, membrane potential, ATP concentration, protein kinase activity, and protease activity
"In this paper, we wanted to demonstrate that our Nano-lanterns are suitable for all these three applications," he adds. Specifically, Movies 1, 2 and 3 of peroxisomes, endosomes and focal adhesions, respectively exemplify the probe's localization and dynamics performance; the Wnt and embryonic stem cell applications illustrate their function as gene expression reporters; and the Ca2+ reporter shows Nano-lanterns as indicators for physical or chemical states. (Wnt – which stands for Wingless-related integration site, after its early nomenclature – signaling pathways are protein structures that pass signals from outside of a cell through the cell surface receptors to the cell interior.)
"It was well known that luminescence imaging is free from photodamage, but luminescence probes were too dim compared to the fluorescent probes. Therefore, luminescence imaging could not be an alternative for the fluorescence imaging – but Nano-lantern has changed the game. The experiments described above correctively demonstrate that our Nano-lantern is suitable for all the major applications of fluorescent probes. Furthermore, as shown with the focal adhesion imaging, Nano-lantern is free from photodamage. In other words, Nano-lantern imaging is now a good alternative for fluorescence imaging when one wants to avoid photodamage."
Long-term continuous luminescence imaging of vinculin-ONL in an MDCKII cell. Images with 1-s exposure were repeated for 240 min. Playback speed is 6,000-fold. (Scale bar: 10 μm.) Credit: Takai A, et al (2015) Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc Natl Acad Sci USA 112(14):4352-4356.
Moving forward, Okada notes, further efforts in developing luminescent protein probes will take three directions: increased brightness, longer wavelength, and improved substrate regeneration and supply. "However," he points out, "our own research interest is in the application of Nano-lanterns for the analysis of gene expression at the single cell level – and monitoring gene expression by Wnt signaling and multicolor monitoring in embryonic stem cells were the first steps towards this application."
In addition, the scientists are developing a so-called super-resolution microscope2. Therefore, super-resolution luminescence imaging is the next step, Okada adds – but one that he nevertheless acknowledges to be a very significant challenge.
Regarding other areas of research that stand to benefit from their Nano-lantern research, Okada tells Phys.org that he is confident that the Nano-lanterns' brightness will be not only beneficial for microscopy imaging, but also for detection with a multiwell plate or cuvette. (A multiwall plate – also known as a microtiter plate, microplate or microwell plate – is a flat plate with multiple microwells used as small test tubes; a cuvette is a small plastic, glass or fused quartz tube designed to hold samples for spectroscopic experiments.) "In fact," he concludes, "luminescent probes based on our Nano-lanterns are bright enough to be detected with a smartphone camera. Expensive detectors such as photomultiplier tubes are therefore unnecessary."
More information: Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging, Proceedings of the National Academy of Sciences (2015) 112(14):4352-4356, doi:10.1073/pnas.1418468112
Related:
1Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%, Nature Communications (2012) 3:751, doi:10.1038/ncomms1738
2Ultrafast superresolution fluorescence imaging with spinning disk confocal microscope optics, Molecular Biology of the Cell, published online before print February 25, 2015, doi:10.1091/mbc.E14-08-1287
Journal information: Proceedings of the National Academy of Sciences , Nature Communications , Molecular Biology of the Cell
© 2015 Phys.org