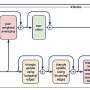
The flow chart above shows how UCSD bioengineers used high-throughput data generated from cells grown under different conditions to form the basis for elucidation of the transcription unit architecture of the E. coli genome. Credit: UC San Diego
Over the last 20 years, the sequencing of the human genome, along with related organisms, has represented one of the largest scientific endeavors in the history of mankind. The information collected from genome sequencing will provide the raw data for the field of bioinformatics, where computer science and biology meet.
Since the publication of the first full genome sequence in the mid-1990s, scientists have been working to identify the genomic location of all the gene products involved in the complex biological processes in a single organism. However, they have only been able to identify a fraction of those locations. Until now. Bioengineers at UC San Diego have made a breakthrough development that will now allow scientists to perform full delineation of the location and use of genomic elements. The researchers have discovered that multiple simultaneous genome-scale measurements are needed to identify all gene products, and to determine their cellular locations and interactions with the genome.
In a recent Nature Biotechnology paper, "The transcription unit architecture of the Escherichia coli genome," the researchers describe a four-step systems approach that integrates multiple genome-scale measurements on the basis of genetic information flow to identify the organizational elements and map them onto the genome sequence. The bioengineers have applied this approach to the E. coli genome to generate a detailed description of its transcrip¬tion unit architecture.
"What's important about this paper is it now enables us to experimentally annotate genomes," said Bernard Palsson, a UCSD bioengineering professor and co-author of the paper. "All this information gives us a fine resolution of the contents of a genome and location of its elements. This is a fine blueprint of a genetic makeup of the genome. We have been able to use genome scale computational models that have been developed at UCSD under the systems biology program, which have enabled us to compute organism designs with higher resolution or better accuracy, which has not been possible before. It takes a lot of the guesswork out of making an organism. Currently there is extensive trial and error in gene sequencing procedures. Hopefully this 'metastucture' of a genome that we have developed will eliminate that trial and error and will enable us to reach new metabolic designs faster with lower failure rates."
Palsson said there are many significant implications of this new finding, such as enhancing metabolic engineering (such as the engineering of microorganisms to make fuels and commodity chemicals).
The UCSD bionengineers combined several computation methods with information mapping in this research. "There are several high throughput methods developed recently like deep sequencing and micro array systems that we used," said Byung-Kwan Cho, a project scientist in the UCSD bioengineering department and the lead author of the Nature Biotechnology paper. "We wanted to integrate all the information into one format to describe the genome. We have genome sequences but we don't know what all of them are. When we sequenced the Human Genome we thought we knew everything but actually we don't know everything. There are lots of data generation techniques and a huge amount of data available. So we were able to map all of this information into one genome sequence.
"So far, scientists have been able to make chemicals to kill pathogenic strains but we haven't been as successful as we have wanted to be," Cho added. "By using this newly discovered information we may be able to design better drugs or medicines to kill pathogenic strains. That's the important point of this research - there is a huge amount of applications for this. The E. coli bacteria is just the beginning."
Source: University of California - San Diego (news : web)