Protein quality control and mitochondria

Protein aggregates are toxic for mitochondrial function, and thus disrupt the supply of chemical energy to their host cells. An LMU team has characterized a protein complex that prevents the build-up of such deposits in the organelles.
Mitochondria are the power stations that supply the cells of higher organisms with the chemical energy necessary for their metabolism and maintenance. In addition, the biosynthesis of many essential metabolites takes place in these organelles. Therefore, any perturbations in their function—such as abnormal accumulations of misfolded proteins—must be rapidly detected and repaired. Research teams led by LMU's Walter Neupert (Biomedical Center) and Roland Beckmann (Gene Center) in cooperation with Toshifumi Inada (Sendai University, Japan) have now elucidated one of the mechanisms by which the cell inhibits the formation of toxic aggregates of proteins destined for the mitochondria, which could otherwise cut off the supply of energy for indispensable cellular functions. The new findings appear in the journal Nature.
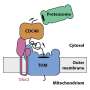
Proteins are synthesized by organelles called ribosomes. These RNA-protein complexes decode the messenger RNA (mRNA) blueprints exported from the nucleus, which specify the amino-acid sequence of the particular protein. Ribosomes successively attach to mRNA molecules at a defined initiation site. This allows each mRNA molecule to program the synthesis of many copies of each particular protein in an assembly-line fashion. If this process is interrupted and the leading ribosome stalls, the following ribosomes pile up behind it. Under these conditions, their attached (incomplete) proteins can readily interact with each other to form protein aggregates. To resolve such traffic jams, cells activate a 'ribosome-associated quality control (RQC) mechanism." The RQC pathway operates by dissociating the two subunits of the stalled ribosomes (thus releasing the mRNA) and attaching the amino acids alanine (A) and threonine (T) to the stalled C-terminal end of the defective protein on the 60S subunit. In this way, the incomplete protein is released from the ribosome and the "CAT tail" marks it for subsequent degradation.
In the case of mitochondrial proteins, however, there is a further complication. Most arrested or stalled mitochondrial proteins are made in the cytoplasm by ribosomes that are attached to pores in the mitochondrial membrane, through which the nascent proteins are directly fed into the organelle. This tight linkage of synthesis with mitochondrial uptake hinders the release and destruction of CAT-tagged proteins in the cytoplasm. Furthermore, CAT-tagged proteins themselves have an increased tendency to aggregate, so their import is especially harmful for mitochondrial function.
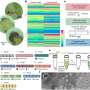
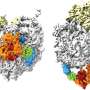
Luckily, the cell has evolved an alternative form of RQC specifically for mitochondrial proteins. The authors of the new study have previously shown that the protein Vms1 plays a key role in this pathway, but the precise mechanism underlying this mode of RQC has remained unclear. "By combining structural analyses based on cryo-electron microscopy with biochemical, molecular biological and genetic experiments, we have now succeeded in elucidating how Vms1 works," says Beckmann. Vms1 is capable of dissociating the ribosome and releasing the stalled protein whether or not the latter bears a CAT sequence. It therefore effectively counteracts the addition of CAT sequences to mitochondrial proteins, thus reducing the risk of aggregation and its deleterious effects on mitochondrial—and consequently on cellular—function. In addition, Beckmann and colleagues identified a previously uncharacterized protein that plays a role in this whole process, and shown how it interacts with Vms1. The new results may contribute to a better understanding of a wide range of disorders, including metabolic diseases and neurodegenerative conditions, which are associated with damage to mitochondrial function.
More information: Ting Su et al. Structure and function of Vms1 and Arb1 in RQC and mitochondrial proteome homeostasis, Nature (2019). DOI: 10.1038/s41586-019-1307-z
Journal information: Nature
Provided by Ludwig Maximilian University of Munich