Controlling gene expression: How chromatin remodelers block a histone pass

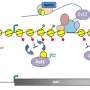
Two opposing teams battle it out to regulate gene expression on the DNA playing field. One, the activators, keeps DNA open to enzymes that transcribe DNA into RNA. Their repressor opponents antagonize that effort by twisting DNA into an inaccessible coil around histone proteins, an amalgam called chromatin, effectively blocking access to DNA by enzymes that elongate an RNA strand.
Both teams maneuver by chemically modifying histones—the activators by decorating histones with acetyl groups—let's call them green flags—causing them to loosen their grip on DNA. The repressors retaliate by marking histones with red flags, often methyl groups, which call in de-acetylase enzymes to clip off the green flags, restore the chromatin barrier and end that round of gene expression. Disturbing this biochemical balance lies at the heart of many diseases, particularly cancer.
Recently, the lab of Jerry Workman, Ph.D., investigator at the Stowers Institute for Medical Research, reported in the journal Nature that a reserve of "pre-acetylated" histones sits on the chromatin sidelines ready to sub for histones whose green flags get clipped by repressors, a tactic aiding the activators called "histone exchange". In a companion study published in the Aug. 26, 2012 Advance Online Publication of Nature Structural & Molecular Biology the Workman lab now shows that a repressor called Set2 in yeast recruits a protein assistant to block the histone exchange. That study reveals a heretofore unknown mechanism to keep gene activation under control and ensure that erroneous transcription does not occur.
"Accurate gene expression is critical for normal cell function, and when this control is lost cells grow abnormally," says Workman. "These two studies identify mechanisms used by cells to regulate gene expression, which is important for our understanding of what goes wrong in diseases marked by unregulated cell growth, like cancer."
The study began when the group, in collaboration with Stowers proteomics experts Michael Washburn, Ph.D., and Laurence Florens, Ph.D., applied mass spectrometry analysis to identify any protein expressed in yeast Saccharomyces cerevisiae that bound to chromatin in regions patrolled by Set2. Those regions were readily apparent by the presence of Set2's red flag methyl group planted in a specific histone protein interacting with DNA.
"We knew that Set2 added this mark in the middle and downstream parts of genes to recruit de-acetylases," says the study's lead author Michaela Smolle, Ph.D., a postdoctoral researcher in the Workman lab. "But the proteomic search allowed us to cast a wide net for other proteins associated with that mark—a bit like fishing."
Among the fish caught was a component of a yeast chromatin "remodeler" known as Isw1, providing circumstantial evidence that the Set2 red flag attracts Isw1 as well as de-acetylases. Additional genomic experiments evaluating the entire genome of a yeast mutant lacking Set2 supported that idea: not only were the red methyl flags missing but the chromatin landscape was devoid of Isw1 as well.
To assess Isw1's biological function the group exploited yet another yeast mutant, this one lacking the ISW1 gene itself. Microarray analysis of global transcription in ISW1 CHD1 mutants showed widely perturbed gene expression marked by aberrant expression of RNA snippets rather than complete transcripts. Biologists view the presence of such "cryptic transcripts" as indicators of cellular stress.
Analysis of acetylation and methylation patterns in chromatin of ISW1 mutants revealed the probable cause: mutants showed ramped up histone exchange activity marked by excessive levels green-flagged pre-acetylated histones along the length of many genes, a condition likely favoring initiation of truncated RNAs.
"Our work shows that the Set2 methylation mark plays two important roles to ensure that RNA transcription starts only at the beginning of the gene and not in the middle," says Workman. "On one hand, it recruits Isw1 to block incoming histones, and on the other it also recruits a deacetylase to remove any acetylation marks that might happen to have sneaked in."
The Workman lab uses yeast, fruit flies and mammalian cells to study multiple factors that activate and repress transcription. Thus far, they have characterized players in the repressive Set2 pathway primarily in yeast, in part because yeast represents an ideal organism for mutational analysis and has a fairly small genome—6,000 genes compared to the 25,000 or so in humans or mice.
Smolle notes that although yeast genomes are simpler, the principles that govern Set2 activity in yeast will likely hold true for its human counterparts, among them the human protein SETD2. "Yeast has a single methylase, while humans have several, and flies and humans have several Isw1-like proteins," she says. "While you cannot equate one with the other precisely, you can be reasonably sure that what happens in yeast happens in humans as well but tends to be more complicated."
Workman agrees, adding that these discoveries have significant implications for human disease. "Strikingly, the human homolog of Set2, SETD2, is implicated as a tumor suppressor in breast cancer and in renal carcinoma, and those cells are deficient in the SETD2 methylation mark," he says. "These findings strongly suggest that SETD2 could be important in controlling cell growth and preventing tumors. Thanks to yeast, we now know more about how Set2 does that."
Journal information: Nature , Nature Structural & Molecular Biology
Provided by Stowers Institute for Medical Research