March 15, 2023 report
This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
New computational method to identify location of cell types in a sample
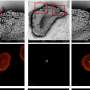
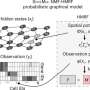
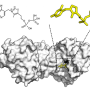
. In contrast, CytoSPACE efficiently assigns individual single-cell transcriptomes to ST coordinates (that is, spots) using global optimization to minimize a correlation-based cost function. This enables downstream analysis of cell type proportions and single-cell transcriptional heterogeneity in spatial dimensions (bottom). The labels \(a_1,...,d_1\) denote individual single cells of cell type \(a,...,d\), respectively, assigned to the featured spot. Credit: Nature Biotechnology (2023). DOI: 10.1038/s41587-023-01697-9")
Stanford University researchers have developed a computational method for identifying where cells are situated in a sample when capturing spatial transcriptomics. The method combines data from spatial transcriptions and a reference single-cell RNA atlas to create modeling outputs. The resulting models can be used to view cellular substructures, identify colocalization patterns and analyze differential expression within a cell type by location.
The new method—cellular Spatial Positioning Analysis via Constrained Expression alignment (CytoSPACE)—has been published in the journal Nature Biotechnology. The script to run the computation has been made freely available via GitHub.
Medical research is very interested in understanding interactions within and between cells. While the genome has taught us a lot about genetic links to disease, the expression of genes in different tissues varies widely. The role of localized cell tissues in preventing or manifesting disease is critical to understanding, predicting, diagnosing and treating an enormous range of diseases.
Cells within tissues communicate with each other, sending group or individual chemical messages, micro-managing the healthy operation of cellular functions. In the case of cancer, it is often a breakdown in communication from within a single cell that can lead to the disease forming as tumorous cells lose the ability to listen to the group's instructions to stop growing and self-destruct.
Single-cell RNA sequencing can capture gene expression (RNA molecules) in individual cells with a high resolution allowing for comparison with other cells. This technique starts with isolating single cells from the tissue of interest. While it tells a researcher everything about expressed genes within the individual cell sequenced, it says nothing about the cells around it.
Current methods of spatial transcriptomics take a more geographical overview of gene expression to create a map of cellular relationships. Instead of individual cell information, researchers get a sampling of ten or more cells contributing to a location. Essentially, a tissue sample is placed against an RNA capture slide that attaches barcodes to the RNA. When the RNA is removed from the slide and sequenced, the barcode can be traced back to its collection location on the slide, and therefore the spatial arrangement of the sequenced reads can be reconstructed into a map.
In the search for more accurate and precise data, several computational methods have been developed to infer general cellular composition in spatial transcriptomic samples. CytoSPACE utilizes single-cell data from a reference scRNA-seq atlas and positions them in a higher resolution.
CytoSPACE constructs a putative set of input single-cell sequences matching what is predicted to be present in the spatial transcriptomics sample. It then infills those sequences to a generated set of locations according to the predicted cellular density of each spatial read. With these matched sets, CytoSPACE takes on the tissue reconstruction task as a linear assignment problem and optimally maps individual cells in scRNA-seq data to spatial coordinates.
The current study by the research group that developed CytoSPACE tested it against the several leading computational methods available. According to the paper, "Across diverse platforms and tissue types, we show that CytoSPACE outperforms previous methods with respect to noise tolerance and accuracy, enabling tissue cartography at single-cell resolution."
More information: Milad R. Vahid et al, High-resolution alignment of single-cell and spatial transcriptomes with CytoSPACE, Nature Biotechnology (2023). DOI: 10.1038/s41587-023-01697-9
Journal information: Nature Biotechnology
© 2023 Science X Network