Biologists glean insight into repetitive protein sequences
. DOI: 10.7554/eLife.77058")
About 70% of all human proteins include at least one sequence consisting of a single amino acid repeated many times, with a few other amino acids sprinkled in. These "low-complexity regions" are also found in most other organisms.
The proteins that contain these sequences have many different functions, but MIT biologists have now come up with a way to identify and study them as a unified group. Their technique allows them to analyze similarities and differences between LCRs from different species, and helps them to determine the functions of these sequences and the proteins in which they are found.
Using their technique, the researchers have analyzed all of the proteins found in eight different species, from bacteria to humans. They found that while LCRs can vary between proteins and species, they often share a similar role—helping the protein in which they're found to join a larger-scale assembly such as the nucleolus, an organelle found in nearly all human cells.
"Instead of looking at specific LCRs and their functions, which might seem separate because they're involved in different processes, our broader approach allows us to see similarities between their properties, suggesting that maybe the functions of LCRs aren't so disparate after all," says Byron Lee, an MIT graduate student.
The researchers also found some differences between LCRs of different species and showed that these species-specific LCR sequences correspond to species-specific functions, such as forming plant cell walls.
Lee and graduate student Nima Jaberi-Lashkari are the lead authors of the study, which appears today in eLife. Eliezer Calo, an assistant professor of biology at MIT, is the senior author of the paper.
Large-scale study
Previous research has revealed that LCRs are involved in a variety of cellular processes, including cell adhesion and DNA binding. These LCRs are often rich in a single amino acid such as alanine, lysine, or glutamic acid.
Finding these sequences and then studying their functions individually is a time-consuming process, so the MIT team decided to use bioinformatics—an approach that uses computational methods to analyze large sets of biological data—to evaluate them as a larger group.
"What we wanted to do is take a step back and instead of looking at individual LCRs, to try to take a look at all of them and to see if we could observe some patterns on a larger scale that might help us figure out what the ones that have assigned functions are doing, and also help us learn a bit about what the ones that don't have assigned functions are doing," Jaberi-Lashkari says.
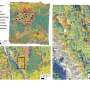
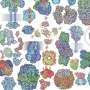
To do that, the researchers used a technique called dotplot matrix, which is a way to visually represent amino acid sequences, to generate images of each protein under study. They then used computational image processing methods to compare thousands of these matrices at the same time.
Using this technique, the researchers were able to categorize LCRs based on which amino acids were most frequently repeated in the LCR. They also grouped LCR-containing proteins by the number of copies of each LCR type found in the protein. Analyzing these traits helped the researchers to learn more about the functions of these LCRs.
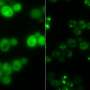
As one demonstration, the researchers picked out a human protein, known as RPA43, that has three lysine-rich LCRs. This protein is one of many subunits that make up an enzyme called RNA polymerase 1, which synthesizes ribosomal RNA. The researchers found that the copy number of lysine-rich LCRs is important for helping the protein integrate into the nucleolus, the organelle responsible for synthesizing ribosomes.
Biological assemblies
In a comparison of the proteins found in eight different species, the researchers found that some LCR types are highly conserved between species, meaning that the sequences have changed very little over evolutionary timescales. These sequences tend to be found in proteins and cell structures that are also highly conserved, such as the nucleolus.
"These sequences seem to be important for the assembly of certain parts of the nucleolus," Lee says. "Some of the principles that are known to be important for higher order assembly seem to be at play because the copy number, which might control how many interactions a protein can make, is important for the protein to integrate into that compartment."
The researchers also found differences between LCRs seen in two different types of proteins that are involved in nucleolus assembly. They discovered that a nucleolar protein known as TCOF contains many glutamine-rich LCRs that can help scaffold the formation of assemblies, while nucleolar proteins with only a few of these glutamic acid-rich LCRs could be recruited as clients (proteins that interact with the scaffold).
Another structure that appears to have many conserved LCRs is the nuclear speckle, which is found inside the cell nucleus. The researchers also found many similarities between LCRs that are involved in forming larger-scale assemblies such as the extracellular matrix, a network of molecules that provides structural support to cells in plants and animals.
The research team also found examples of structures with LCRs that seem to have diverged between species. For example, plants have distinctive LCR sequences in the proteins that they use to scaffold their cell walls, and these LCRs are not seen in other types of organisms.
The researchers now plan to expand their LCR analysis to additional species.
"There's so much to explore, because we can expand this map to essentially any species," Lee says. "That gives us the opportunity and the framework to identify new biological assemblies."
More information: Byron Lee et al, A unified view of low complexity regions (LCRs) across species, eLife (2022). DOI: 10.7554/eLife.77058
Journal information: eLife
Provided by Massachusetts Institute of Technology
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.