How large-scale single cell genomics complements metagenomics studies

Researchers demonstrated the value of conducting large-scale single cell genomics by collecting nearly 500 single cells from a single low diversity hot spring sediment sample. Their work showed that single cell genomics can add significant value to the other commonly used culture-independent sequencing approaches including amplicon and metagenomic sequencing. For example, they showed that the composition of the community was similar across sequencing approaches, that species specific sets of single cells harbored mobile genetic elements that were missed within paired metagenome assembled genomes (MAGs), and that dominant populations varied with respect to the amount of within species recombination, indicating variation in gene flow between the analyzed community members.
Though microbes help regulate the planet's nutrient cycles and are potentially of use in fields ranging from agriculture to biotechnology and medicine, the vast majority in, on, and around the planet remain unknown. In recent years, advances in sequencing technologies and bioinformatic tools have helped decode the genomes of tens of thousands of previously unknown and uncultivated microbes through metagenomics. Such techniques take advantage of bioinformatics tools for extracting snippets of microbial genomes directly from environmental sequence data, by piecing each genome together from large mixtures of genomic sequences. A complementary approach to this is single cell genomics, where cells from environmental samples are first separated, and their genomes then amplified and sequenced individually, offering scientists the opportunity to apply population genomics approaches to closely related cells plucked directly from the environment.
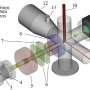
Dewar Creek is a remote hot spring, deep within the British Columbia backcountry (Purcell Wilderness Conservancy Provincial Park of British Columbia, BC Parks). In these springs, temperatures can reach as high as 80C (~ 190F), yet microbes thrive here. The communities in this extreme environment are often less diverse than those within more moderate ecosystems. A few years ago, a candidate bacterial lineage was identified from microbial and metagenome sequence data sets generated from a handful of hot springs, including Dewar Creek.
Continuing explorations into this unique environment, researchers from the University of Calgary and the U.S. Department of Energy (DOE) Joint Genome Institute (JGI), a DOE Office of Science User Facility located at Lawrence Berkeley National Laboratory (Berkeley Lab), employed single-cell sequencing to assess the diversity within and between microbial populations. The work appeared in The ISME Journal.
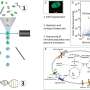
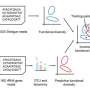
Members of the Dunfield lab collected samples from this hot spring. A single sample was then used to produce a paired amplicon dataset (a.k.a. 16S rRNA gene sequencing), a shotgun metagenome, and a single cell dataset of nearly 500 single cell genomes. The single cell portion of this work was performed by Danielle Goudeau and Rex Malmstrom of the JGI's Microscale Applications group. Cells were randomly sorted, whole genome amplified, then sequenced and assembled. Robert Bowers, a JGI scientist within the Microbial Program, led the genomic analyses to compare the resulting datasets, emphasizing the utility of single cell sequencing to assess the variation within natural microbial populations.
Each of the three sequencing approaches produced a generally similar community profile. But each approach demonstrates its full value at different scales. Amplicon sequencing is commonly used to assess fluctuations in microbial diversity across thousands of samples. Metagenomics is currently being applied across tens to hundreds of samples, while single cell approaches have been typically used as a complement to isolate sequencing, i.e., when the targeted cells cannot be cultivated in the lab.
What makes this particular study unique is the application of single cell genomics to a whole community. Given the low microbial diversity of the sampled hot spring, a dataset of nearly 500 single cells covered the diversity of most taxa within the sample. Furthermore, the three most abundant lineages were represented by enough single cell genomes to facilitate an analysis of within and between population heterogeneity by comparing the ATGCs of the genomes from each single cell. The team showed that while the broad nucleotide-level diversity was similar across the dominant lineages, each microbial group displayed vastly different recombination profiles. This is akin to the structure of social media networks where one social media group may have a relatively constrained set of friends, while another might exhibit few limitations to interactions and new connections, thus sharing more ideas, similar to the sharing of genes within a highly recombining microbial population. This work showcases the utility of single cell sequencing, as monitoring population-level heterogeneity of uncultivated microbes will provide researchers the ability to capture the fine-scale variation within populations that is a precursor to strain-level diversification and microbial speciation.
More information: Robert M. Bowers et al, Dissecting the dominant hot spring microbial populations based on community-wide sampling at single-cell genomic resolution, The ISME Journal (2021). DOI: 10.1038/s41396-021-01178-4
Journal information: ISME Journal
Provided by DOE/Joint Genome Institute