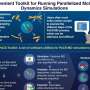
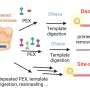
How to target a microbial needle within a community haystack
. 2. These targeted cells are enriched based on their FISH signal via flow cytometric cell sorting. 3. The enriched population is then sequenced and further analyzed. Genome annotation reveals putative functional repertoires. Credit: Overview and Figure 1: Courtesy of Anissa Grieb, Figures 2 and 3: Grieb A et al. A pipeline for targeted metagenomics of environmental bacteria. Microbiome. 8:21;2020 Feb 17, Figures 4 and 6, http://creativecommons.org/licenses/by/4.0/)")
A team led by researchers at the Max Planck Institute (MPI) for Marine Microbiology has developed, tested and deployed a pipeline to first target cells from communities of uncultivated microbes, and then efficiently retrieve and characterize their genomes.
Despite great strides in generating genome sequences for members of the microbial world through bulk community sequencing (metagenomics) and single-cell genomics, the vast majority of microbes that play key roles in global biogeochemical cycles remains unexplored and uncultivated. Developing a more targeted genomic approach would allow researchers to focus on specific microbes within metagenome samples and associate their sequences with their roles within their environments.
The traditional shotgun metagenomics approach to learning more about environmental samples starts with researchers first sequencing the complex community and then separating out the sequences representing microbes of interest. When the microbe of interest is a tiny fraction of that complex community, the search for its genome amidst all the other sequences can be even more challenging.
To overcome this hurdle, a team led by Bernhard Fuchs and his postdoctoral researcher Anissa Grieb at the MPI for Marine Microbiology worked with researchers at the U.S. Department of Energy (DOE) Joint Genome Institute (JGI), a DOE Office of Science User Facility located at Lawrence Berkeley National Laboratory (Berkeley Lab) to develop a process for more easily identifying cells of interest in a complex environmental sample and then sorting these cells so that their DNA could be sequenced. Their protocol, detailed in the journal Microbiome, combines multiple techniques and was demonstrated using a specific group of marine bacteria called "Vis6" that are found in North Sea algal blooms.
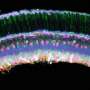
The workflow starts by tagging specific cells using an approach named hybridization chain reaction fluorescence in situ hybridization (HCR-FISH). In contrast to standard FISH where a one probe adds one fluorescent molecule when bound to a target, HCR-FISH attaches multiple fluorescent tags to a single binding site, resulting in target cells that give off a much brighter signal. Cells from many natural environments are typically too dim to see by flow cytometry when using standard FISH probes, so the signal amplification provided by HCR-FISH is critical for detection. Once cells are labeled with HCR-FISH, they can be collected using fluorescence activated cell sorting (FACS) into smaller, less diverse pools before being subjected to genome sequencing. Various fixation protocols were also tested as part of the method development, and researchers found that ethanol treatment improved fluorescence signals without hampering subsequent DNA amplification and sequencing steps.
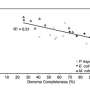
Testing the HCR-FISH+FACS targeted pipeline on North Sea metagenomes as a proof of principle, the team used a Vis6-specific probe and was able to amplify the Vis6 group, which accounted for less than one percent of the sequences from a bulk metagenome, to just over 50 percent of the targeted metagenome sequence reads. This demonstrates the viability of HCR-FISH+FACS for selectively enriching low abundance microbial groups from complex natural communities. This type of enrichment could be applied in a number of ways including gathering a more complete view of a population's genetic makeup and identifying viruses interacting with these populations.
Development of this protocol was enabled by the JGI's Emerging Technologies Opportunity Program. The ETOP helps researchers develop technology that can then be made available to JGI users in order to address pressing energy and environmental scientific challenges.
More information: Anissa Grieb et al. A pipeline for targeted metagenomics of environmental bacteria, Microbiome (2020). DOI: 10.1186/s40168-020-0790-7
Provided by DOE/Joint Genome Institute