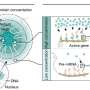
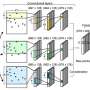
Machine learning algorithm predicts how genes are regulated in individual cells
 and existing data on transcription factor binding sites (\"ChIP-seq TF-Target gene matrix\") are analyzed to predict transcription factor activity (\"Inferred TF activity\") that can be leveraged for a broad range of analyses. Credit: Genome Research, Attribution 4.0 International CC BY 4.0 license")
A team of scientists at the University of Illinois Chicago has developed a software tool that can help researchers more efficiently identify the regulators of genes. The system leverages a machine learning algorithm to predict which transcription factors are most likely to be active in individual cells.
Transcription factors are proteins that bind to DNA and control what genes are turned "on" or "off" inside a cell. These proteins are relevant to biomedical researchers because understanding and manipulating these signals in the cell can be an effective way to discover new treatments for some illnesses. However, there are hundreds of transcription factors inside human cells and it can take years of research, often through trial and error, to identify which are most active—those that are expressed, or "on"—in different types of cells and that could be leveraged as drug targets.
"One of the challenges in the field is that the same genes may be turned 'on' in one group of cells but turned 'off' in a different group of cells within the same organ," said Jalees Rehman, UIC professor in the department of medicine and the department of pharmacology and regenerative medicine at the College of Medicine. "Being able to understand the activity of transcription factors in individual cells would allow researchers to study activity profiles in all the major cell types of major organs such as the heart, brain or lungs."
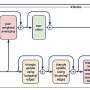
Named BITFAM, for Bayesian Inference Transcription Factor Activity Model, the UIC-developed system works by combining new gene expression profile data gathered from single cell RNA sequencing with existing biological data on transcription factor target genes. With this information, the system runs numerous computer-based simulations to find the optimal fit and predict the activity of each transcription factor in the cell.
The UIC researchers, co-led by Rehman and Yang Dai, UIC associate professor in the department of bioengineering at the College of Medicine and the College of Engineering, tested the system in cells from lung, heart and brain tissue. Information on the model and the results of their tests are reported today in the journal Genome Research.
"Our approach not only identifies meaningful transcription factor activities but also provides valuable insights into underlying transcription factor regulatory mechanisms," said Shang Gao, first author of the study and a doctoral student in the department of bioengineering. "For example, if 80% of a specific transcription factor's targets are turned on inside the cell, that tells us that its activity is high. By providing data like this for every transcription factor in the cell, the model can give researchers a good idea of which ones to look at first when exploring new drug targets to work on that type of cell."
The researchers say that the new system is publicly available and could be applied widely because users have the flexibility to combine it with additional analysis methods that may be best suited for their studies, such as finding new drug targets.
"This new approach could be used to develop key biological hypotheses regarding the regulatory transcription factors in cells related to a broad range of scientific hypotheses and topics. It will allow us to derive insights into the biological functions of cells from many tissues," Dai said.
Rehman, whose research focuses on the mechanisms of inflammation in vascular systems, says an application relevant to his lab is to use the new system to focus on the transcription factors that drive diseases in specific cell types.
"For example, we would like to understand if there is transcription factor activity that distinguished a healthy immune cell response from an unhealthy one, as in the case of conditions such as COVID-19, heart disease or Alzheimer's disease where there is often an imbalance between healthy and unhealthy immune responses," he said.
More information: Shang Gao et al, A Bayesian inference transcription factor activity model for the analysis of single-cell transcriptomes, Genome Research (2021). DOI: 10.1101/gr.265595.120
Journal information: Genome Research
Provided by University of Illinois at Chicago