Statistical methods improve biological single-cell analyses

Stem cells can turn into heart cells, skin cells can mutate to cancer cells; even cells of the same tissue type exhibit small heterogeneities. Scientists use single-cell analysis to investigate these heterogeneities. But the method is still laborious and considerable inaccuracies conceal smaller effects. Scientists at the Technische Universitaet Muenchen (TUM), the Helmholtz Zentrum Muenchen and the University of Virginia (USA) have now found a way to simplify and improve the analysis by mathematical methods.
Each cell in our body is unique. Even cells of the same tissue type that look identical under the microscope differ slightly from each other. To understand how a heart cell can develop from a stem cell, why one beta-cell produces insulin and the other does not, or why a normal tissue cell suddenly mutates to a cancer cell, scientists have been targeting the activities of ribonucleic acid, RNA.
Proteins are constantly being assembled and disassembled in the cell. RNA molecules read blueprints for proteins from the DNA and initiate their production. In the last few years scientists around the world have developed sequencing methods that are capable of detecting all active RNA molecules within a single cell at a certain time.
At the end of December 2013 the journal Nature Methods declared single-cell sequencing the "Method of the Year." However, analysis of individual cells is extremely complex, and the handling of the cells generates errors and inaccuracies. Smaller differences in gene regulation can be overwhelmed by the statistical "noise."
Easier and more accurate, thanks to statistics
Scientists led by Professor Fabian Theis, Chair of Mathematical modeling of biological systems at the Technische Universitaet Muenchen and director of the Institute of Computational Biology at the Helmholtz Zentrum Muenchen, have now found a way to considerably improve single-cell analysis by applying methods of mathematical statistics.
Instead of just one cell, they took 16-80 samples with ten cells each. "A sample of ten cells is much easier to handle," says Professor Theis. "With ten times the amount of cell material, the influences of ambient conditions can be markedly suppressed." However, cells with different properties are then distributed randomly on the samples. Therefore Theis's collaborator Christiane Fuchs developed statistical methods to still identify the single-cell properties in the mixture of signals.
Combining model and experiment
On the basis of known biological data, Theis and Fuchs modeled the distribution for the case of genes that exhibit two well-defined regulatory states. Together with biologists Kevin Janes and Sameer Bajikar at the University of Virginia in Charlottesville (USA), they were able to prove experimentally that with the help of statistical methods samples containing ten cells deliver results of higher accuracy than can be achieved through analysis of the same number of single cell samples.
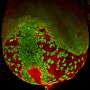
In many cases, several gene actions are triggered by the same factor. Even in such cases, the statistical method can be applied successfully. Fluorescent markers indicate the gene activities. The result is a mosaic, which again can be checked to spot whether different cells respond differently to the factor.
The method is so sensitive that it even shows one deviation in 40 otherwise identical cells. The fact that this difference actually is an effect and not a random outlier could be proven experimentally.
More information: Parameterizing cell-to-cell regulatory heterogeneities via stochastic transcriptional profiles, Sameer S. Bajikar, Christiane Fuchs, Andreas Roller, Fabian J. Theis, and Kevin A. Janes, PNAS, Early Edition, 21 Januar 2014, DOI: 10.1073/pnas.1311647111
Journal information: Nature Methods , Proceedings of the National Academy of Sciences
Provided by Technical University Munich