New atom-level insight into drug-target residence time
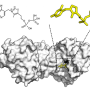
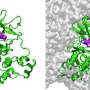
. c Binding modes of 1 and 2 shown with representative snapshots obtained from MD simulations (the snapshots were selected to reflect the observed protein ligand interactions; see d). In the figure that is illustrating the whole kinase, molecular surface of p38α MAPK is displayed in white, and molecular surface for 1 and 2 are shown in light red and light blue, respectively. In the close-up image of the binding site, the key interaction residues are shown with sticks together with solvent molecules in the close proximity of the ligand. Ligands are displayed with ball and stick representations using light red and light blue colors for carbon-atoms of 1 and 2, respectively. H-bonds shown with yellow, π–cation with red and π–π stacking with green dashed lines. d 2D-representation of protein–ligand interaction frequencies. Displayed are the most frequent H-bond, π–cation, and π–π stacking interaction frequencies (interactions with >15% frequency are shown). Source data are provided as a Source Data file. Credit: DOI: 10.1038/s41467-022-28164-4")
A new study from the University of Eastern Finland and the University of Tübingen helps to explain what defines how long a drug molecule stays bound to its target.
When a drug molecule binds to its target protein, it stays bound for some time before eventually unbinding the target. The actual time how long a drug molecule resides bound to its target varies among compounds. The lifetime of the drug-target complex may play a crucial role in drug efficacy, as a long target residence time can, in some cases, be important for drug efficacy. Therefore, understanding its underlying causes enables more rational drug design.
In the new study, researchers from the University of Eastern Finland and the University of Tübingen identified the key elements driving for a long or a short target residence time among similar small molecule kinase inhibitors on the atomistic level. The findings were published in Nature Communications.
Dozens of small molecule kinase inhibitors have already been approved for clinical use, most of them for the treatment of cancer.
"Initially, we were interested in what causes the discrepancy in the target residence time between two similar small molecule kinase inhibitors," says Senior Researcher, lead author Tatu Pantsar from the University of Eastern Finland.
Prof. Stefan Laufer's group at the University of Tübingen has designed, synthesized and biologically characterized numerous small molecule kinase inhibitors for a protein kinase called p38a MAPK, which enabled this research.
"In the study, we focused on two small molecule kinase inhibitors that are equipotent in isolated enzyme assays but show difference in their target residence times, i.e., for how long a single small molecule kinase inhibitor is bound to the target protein. We also found that the inhibitor with a longer target residence time was more potent when tested in cells," Pantsar says.
In the study, researchers studied and compared the small molecule kinase inhibitors together with their target protein using computer simulations that were conducted on Finnish supercomputers.
The protein behaves differently depending on the bound inhibitor.
"The simulations suggest that when a small molecule inhibitor is bound to the protein, the protein is more dynamic when the short residence time inhibitor is bound to it. This basically means that the protein moves more when it is binding the short residence time inhibitor, and less when it is binding the long residence time inhibitor," Pantsar points out.
Water molecules have a great impact on the drug target residence time.
"These tiny yet abundant water molecules surrounding the protein appear to be really important. When the inhibitor unbinds the protein, water molecules will take their place in the binding site."
In the simulations, the long residence time inhibitor was less exposed to the water molecules, and the required energy for the water molecules to reoccupy the binding site of the long residence time inhibitor was much higher. This results in a higher energetical barrier for the inhibitor to dissociate from its target and thereby in a longer lifetime of the drug-target complex.
The observations on target protein behavior and the role of water molecules were also confirmed with a structurally diverse small molecule kinase inhibitor that has an extremely short residence time.
The results can be useful in the early stages of drug design.
"Now that we understand better the atomistic level reasons that define the drug residence time, it enables more rational small molecule design that can be applied in a drug discovery project if a long target residence time is desired. Obviously, one needs to remember that the target residence time is just one aspect in the very complex and difficult drug design process, where a multitude of different things need to be considered," Pantsar concludes.
More information: Tatu Pantsar et al, Decisive role of water and protein dynamics in residence time of p38α MAP kinase inhibitors, Nature Communications (2022). DOI: 10.1038/s41467-022-28164-4
Journal information: Nature Communications
Provided by University of Eastern Finland