Researchers develop method to dramatically reduce error rate in next-generation sequencing
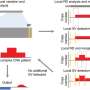
 with corresponding Jinghui Zhang, Ph.D., illustrates the significantly decreased error rate using CleanDeepSeq. Credit: St. Jude Children's Research Hospital")
St. Jude Children's Research Hospital investigators have developed software to shrink the error rate in next-generation sequencing data by as much as 100-fold, which would likely speed early detection of relapse and other threats. The findings appear March 14 in the journal Genome Biology.
Researchers analyzed next-generation DNA sequencing datasets from St. Jude and four other institutions to identify and suppress common sources of sequencing errors. Using the new process, researchers reported that the error rate for DNA base substitution declined from 0.1 percent (1 in 1,000) to between 0.01 (1 in 10,000) and 0.001 percent (1 in 100,000).
By making it easier to distinguish with greater accuracy the signal from noises, in this case a true mutation from a sequencing error, researchers hope to give patients a head start on cures.
"Early detection of cancer or cancer relapse really is like finding a needle in a haystack because the number of cancer cells is overwhelmed by the number of normal cells at early stage," said co-first and corresponding author Xiaotu Ma, Ph.D., an assistant member of the St. Jude Department of Computational Biology. "This method, which we named CleanDeepSeq, helps eliminate the hay to make it easier to find the needle."
Roadblock
Sequencing the human genome involves determining the exact order of the 3 billion chemical bases or letters that make up the genome. DNA base substitutions are the most abundant mutations in children and adults with cancer.
Interest in reducing errors and improving data quality has grown as next-generation sequencing costs have fallen. Massively parallel processing means cancer-driving genes can now be sequenced thousands or hundreds of thousands of times to find clues of cancer cells long before the overt disease.
"Sequencing errors are a roadblock to detecting the low-frequency genetic variants that are important for cancer molecular diagnosis, treatment and surveillance using deep next-generation sequencing," said corresponding and senior author Jinghui Zhang, Ph.D., St. Jude Computational Biology chair. "This study provides the first comprehensive analysis of the source of such sequencing errors and offers new strategies for improving the accuracy."
Error suppression
This study focused on identifying the variety and source of substitution errors in next-generation sequencing data and creating a mathematical error-suppression strategy. Investigators used a variety of techniques to determine the lowest frequency at which a true mutation could be distinguished from a sequencing error. The research involved analyzing datasets from St. Jude, HudsonAlpha Institute of Biotechnology, the Broad Institute, Baylor College of Medicine, and WuXiNextCODE, in China.
The analysis revealed several sources of errors, including handling and storage of the patient samples, the enzymes used to amplify patient samples and the sequencing itself. The profiling led Ma and his colleagues to home in on recognition and suppression of errors related to poor sequencing quality or difficulty re-assembling (mapping) the sequences or aligning the patient genome with a reference genome.
Researchers are working to bring CleanDeepSeq to the clinic for monitoring relapse and possibly early diagnosis, especially in high-risk patients. "This method might also help scientists studying infectious diseases like influenza and HIV or wherever drug-resistance is a concern," Ma said.
More information: Xiaotu Ma et al. Analysis of error profiles in deep next-generation sequencing data, Genome Biology (2019). DOI: 10.1186/s13059-019-1659-6
Journal information: Genome Biology
Provided by St. Jude Children's Research Hospital