This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
The who's who of bacteria: A reliable way to define species and strains
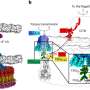
 or all samples combined (Panel A) of the control pond were mapped to the Sal. ruber genomes preserving all matches with identity ≥99.3%. The mapping file was manipulated to remove one target genome at a time (randomly sorted) while recording the number of unique reads mapping at each step, and this process was repeated 100 times to reduce the impact of randomization on the estimates obtained (below). The number of reads were then expressed as the fraction of the maximum number of reads from the Sal. ruber species by dividing the observed counts by the total number of reads mapping to any reference genome with identity ≥ 95%. The logarithm of the number of total (dereplicated) genomes used was then expressed as a function of the fraction of Sal. ruber reads captured by the genomes, and a linear regression was determined by unweighted least squares and evaluated using Pearson correlation for the region between 20 and 100 genomes. This trendline was extrapolated to 100% coverage of the genomovar diversity (i.e., all reads from the species) to provide an estimate of the number of genomovars represented (Y-axis) in the total sequenced fraction (X-axis). Filled dots represent the fraction of the total Sal. ruber reads captured by the genomovars used, and the shaded bands around the observed subsamples represent the central inter-quantile ranges at 100%, 80%, 60, 40%, and 20%. Source data are provided as Source Data 2. Credit: Nature Communications (2024). DOI: 10.1038/s41467-023-44622-z")
What's in a name? A lot, actually. For the scientific community, names and labels help organize the world's organisms so they can be identified, studied, and regulated. But for bacteria, there has never been a reliable method to organize them into species and strains cohesively. It's a problem because bacteria are one of the most prevalent life forms, making up roughly 75% of all living species on Earth.
An international research team sought to overcome this challenge, which has long plagued scientists who study bacteria. Kostas Konstantinidis, Richard C. Tucker Professor in the School of Civil and Environmental Engineering at the Georgia Institute of Technology, co-led a study to investigate natural divisions in bacteria with a goal of determining a scientifically viable method for organizing them into species and strains. To do this, the researchers let the data show them the way.
Their research was published in the journal Nature Communications.
"While there is a working definition for species and strains, this is far from widely accepted in the scientific community," Konstantinidis said. "This is because those classifications are based on humans' standards that do not necessarily translate well to the patterns we see in the natural environment."
For instance, he said, "If we were to classify primates using the same standards that are used to classify E. coli, then all primates—from lemurs to humans to chimpanzees—would belong to a single species."
There are many reasons why a comprehensive organizing system has been hard to devise, but it often comes down to who gets the most attention and why. More scientific attention generally leads to those bacteria becoming more narrowly defined. For example, bacteria species that contain toxic strains have been extensively studied because of their associations with disease and health.
This has been out of the necessity to differentiate harmful strains from harmless ones. Recent discoveries have shown, however, that even defining types of bacteria by their toxicity is unreliable.
"Despite the obvious cornerstone importance of the concepts of species and strains for microbiology, these remain, nonetheless, ill-defined and confusing," Konstantinidis said.
The research team collected bacteria from two salterns in Spain. Salterns are built structures in which seawater evaporates to form salt for consumption. They harbor diverse communities of microorganisms and are ideal locations to study bacteria in their natural environment. This is important for understanding diversity in populations because bacteria often undergo genetic changes when exposed to lab environments.
The team recovered and sequenced 138 random isolates of Salinibacter ruber bacteria from these salterns. To identify natural gaps in genetic diversity, the researchers then compared the isolates against themselves using a measurement known as average nucleotide identity (ANI)—a concept Konstantinidis developed early in his career. ANI is a robust measure of relatedness between any two genomes and is used to study relatedness among microorganisms and viruses, as well as animals. For instance, the ANI between humans and chimpanzees is about 98.7%.
The analysis confirmed the team's previous observations that microbial species do exist and could be reliably described using ANI. They found that members of the same species of bacteria showed genetic relatedness typically ranging from 96 to 100% on the ANI scale and generally less than 85% relatedness with members of other species.
The data revealed a natural gap in ANI values around 99.5% ANI within the Salinibacter ruber species that could be used to differentiate the species into its various strains. In a companion paper published in mBio, the team examined about 300 additional bacterial species based on 18,000 genomes that had been recently sequenced and became available in public databases. They observed similar diversity patterns in more than 95% of the species.
"We think this work expands the molecular toolbox for accurately describing important units of diversity at the species level and within species, and we believe it will benefit future microdiversity studies across clinical and environmental settings," Konstantinidis said.
The team expects their research will be of interest to any professional working with bacteria, including evolutionary biologists, taxonomists, ecologists, environmental engineers, clinicians, bioinformaticians, regulatory agencies, and others. It is available online through Konstantinidis' website and GitHub to facilitate access and use by scientific and regulatory communities.
"We hope that these communities will embrace the new results and methodologies for the more robust and reliable identification of species and strains they offer, compared to the current practice," Konstantinidis said.
More information: Tomeu Viver et al, Towards estimating the number of strains that make up a natural bacterial population, Nature Communications (2024). DOI: 10.1038/s41467-023-44622-z
Luis M. Rodriguez-R et al, An ANI gap within bacterial species that advances the definitions of intra-species units, mBio (2023). DOI: 10.1128/mbio.02696-23
Journal information: Nature Communications , mBio
Provided by Georgia Institute of Technology