Simulations provide map to treasure trove of fluorinated compounds
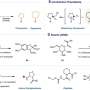
 Reactions between difluorocarbene and numerous pairs of small molecules were simulated, predicting a N-heterocycle product fluorinated twice at the alpha carbon. (Right) The successful reaction framework using pyridine and examples of the types of product compounds obtained. Credit: Nature Synthesis (2022). DOI: 10.1038/s44160-022-00128-y")
Computer simulations are most often used as a guide so chemists can more efficiently work out the exact details of a general reaction idea they have in mind—much like a compass helps guide an explorer efficiently to a destination on their map. However, researchers at ICReDD took things a big step further and used simulations to produce the general idea for an entirely unimagined reaction, effectively using computations to make the map itself. Using the design principle suggested by computational results, the team hit the motherlode in the lab, successfully developing a suite of 48 reactions that produce compounds potentially useful for novel drug development.
The presence and position of fluorine in a molecule often affects a molecule's pharmacological activity. Researchers at ICReDD have utilized quantum chemical calculations to discover a reaction that selectively adds two fluorine atoms to a difficult-to-access position on an N-heterocycle—molecules with a carbon ring structure where at least one carbon in the ring is replaced with nitrogen. The ability to attach fluorine atoms to the previously difficult-to-access "alpha carbon"—the carbon immediately next to the nitrogen in the ring structure—could lead to the development of a host of novel drugs.
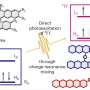
Before carrying out experiments in the lab, the researchers cast a wide net, computationally testing the viability of numerous 3-component reactions using the artificial force induced reaction (AFIR) method. They simulated the reaction of a difluorocarbene molecule, which acts at the source of fluorine atoms, with various pairs of small molecules featuring a double or triple bond. These simulations showed that a number of ring-forming reactions should be viable.
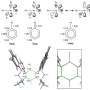
Researchers tried one of the promising reactions suggested by initial computational results but weren't successful. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis.
More information: Satoshi Maeda, In silico reaction screening with difluorocarbene for N-difluoroalkylative dearomatization of pyridines, Nature Synthesis (2022). DOI: 10.1038/s44160-022-00128-y. www.nature.com/articles/s44160-022-00128-y
Journal information: Nature Synthesis
Provided by Hokkaido University