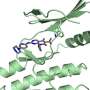
Computer simulations of proteins help unravel why chemotherapy resistance occurs
 bound to the anti-cancer drug imatinib. Normally, the drug exits slowing via the blue arrow. A modification in the kinase (red sphere) causes the drug to exit via a fast route (red arrow). Credit: Aziz M. Rangwala")
Understanding why and how chemotherapy resistance occurs is a major step toward optimizing treatments for cancer. A team of scientists including Markus Seeliger, Ph.D., of the Stony Brook Cancer Center and Renaissance School of Medicine at Stony Brook University, believe they have found a new process through which drug resistance happens. They are using a computer simulation model that is helping them understand exactly how molecules interact with the cancer drug Imatinib (known as Gleevec) in the chemotherapy resistant process. Imatinib treats chronic myeloid leukemia (CML) highly effectively, yet many late stage patients experience drug resistance, which renders the drug minimally effective at that stage.
The research is highlighted in a paper published in Angewandte Chemie and builds upon previous research detailed in 2021 in PNAS.
Imatinib inhibits the BCR-Abl protein kinase, an overly active cellular signaling machinery in CML. In the PNAS study, researchers showed that variations in the building plan of the kinase can make it harder for Imatinib to bind to the kinase and also speed up drug release from the kinase. In the Angewandte Chemie paper, the research team took the computational methodology—developed by co-author Pratyush Tiwary from the University of Maryland—that enabled them to study the very slow release of Imatinib from the kinase.
"This method in itself is a major technical achievement that extends computational abilities for drug resistance research, and importantly led to us being able to predict how rapidly healthy and mutant proteins would release this drug," says Seeliger, Associate Professor in the Department of Pharmacological Sciences. "For the first time, we could see the release of a drug from a protein in such detail and accuracy. Moreover, we could show that the mutation changes fundamentally within the exit route of the drug from the protein.
"This is important since the speed of the drug release may be just as important for the therapeutic effect of a drug as how tightly a drug binds to the protein."
Seeliger further explains that the method could provide a foundation for understanding the molecular mechanisms behind chemotherapy resistance.
More broadly, the implications of what they discovered are that if scientists can understand how drugs are released from their proteins, they may be able to design drugs with a slower release and higher therapeutic impact. Additionally, if rapid drug release could cause drug resistance, and clinicians can show this is happening, they may be able to re-activate the drug effectiveness by asking the patient to take the drug more frequently.
The groundwork for the mutation testing via the computational method was outlined in the PNAS paper. Seeliger and colleagues tested how imatinib binds to mutations in patients with imatinib-resistant CML. They found that the majority of mutations readily bind to imatinib, so that posed the question just how do these mutations cause resistance in patients? The researchers then identified several mutants which bound imatinib readily but they release the drug much faster.
After identifying these mutants with a faster drug release, the team used nuclear magnetic resonance (NMR) and molecular dynamics to link the protein to drug disassociation—underlying the importance of drug disassociation kinetics for drug efficacy. This enabled them to identity a novel mechanism of imatinib resistance.
The work resulting in the paper published in PNAS involved the collaborative efforts of Seeliger and his colleagues at Stony Brook, and researchers at Memorial Sloan Kettering Cancer Center and at Goethe University of Frankfurt, Germany.
Research that resulted in the more recent paper was led by Tiwary and colleagues at the University of Maryland, in collaboration with Seeliger and scientists at the Broad Institute at MIT and Harvard University.
More information: Mrinal Shekhar et al, Protein Flexibility and Dissociation Pathway Differentiation Can Explain Onset of Resistance Mutations in Kinases, Angewandte Chemie International Edition (2022). DOI: 10.1002/anie.202200983
Agatha Lyczek et al, Mutation in Abl kinase with altered drug-binding kinetics indicates a novel mechanism of imatinib resistance, Proceedings of the National Academy of Sciences (2021). DOI: 10.1073/pnas.2111451118
Journal information: Proceedings of the National Academy of Sciences , Angewandte Chemie , Angewandte Chemie International Edition
Provided by Stony Brook University