'Hotspot mapping' accelerates early-phase drug design

The amount of structural data on protein drug targets continues to grow. However, successfully mining this data to form testable hypotheses that drive drug discovery can prove challenging. Selectivity for the target protein is a crucial property in the development of new therapeutics. In a recent paper in the Journal of Chemical Information and Modeling, authors from the CCDC, Exscientia, and Oxford University show how an automated process leveraging "ensemble hotspot maps" can identify key structural differences that contribute to the selectivity of a compound for one protein over another.
The power of hotspot mapping to advance drug design
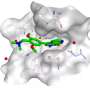
Hotspot mapping quantifies the propensity for compounds to exploit interactions in a preferred binding site—providing a 3-D grid of data to help score and prioritize compounds. The power of this method lies in how it finds key interactions during early-phase drug discovery and then distills the information into easily interpretable results. Chris Radoux is Head of Structural Bioinformatics at Exscientia and a co-author on the paper.
"Adding hotspot maps early in a drug discovery project can provide a molecular blueprint using the protein structure alone," says Radoux. "This can be used to help determine how druggable a given pocket of a target protein is and to prioritize fragment starting points for compound design. The highest scoring interactions can then be used to guide computational methods and algorithms."
Hotspot maps drive cohesive drug design
This approach automates analysis across a protein family, as proteins in the same family often have similar binding sites. According to Mihaela D Smilova, co-author and postgraduate researcher at the Centre for Medicines Discovery at Oxford University, selectivity profiles in both the complete proteome—and within the target protein family—must be understood to develop safe and effective drugs. Interactions with unrelated target proteins can lead to unwanted side effects and toxicity, she says. However, effective drugs often exploit the benefits of "polypharmacology."
"Introducing polypharmacology, or the ability to modulate multiple targets, may help to prevent the development of resistant disease phenotypes," says Smilova. "Consequently, a successful drug candidate has a finely tuned selectivity profile within its target family—interacting with targets that positively impact the disease phenotype and avoiding interactions that lead to unwanted side effects."
Using hotspot maps as inputs for computational workflows means researchers can rapidly explore the chemical space.
"This saves time by summarizing the information and presenting it in a way that is both interpretable by medicinal chemists and can be used in further computational analyses," says Smilova.
Leveraging real-world, empirical data for reliability
The script used to generate the hotspot maps is a Python package called "Hotspots API," which leverages the data in the Cambridge Structural Database (CSD) via CCDC's IsoStar library of interactions. The CSD is the world's repository for small-molecule organic and metal-organic crystal structures—containing over 1.1 million structures from X-ray and neutron diffraction analyses. IsoStar is a web application that uses the CSD to generate thousands of interactive 3-D scatterplots that show the probability of occurrence and spatial characteristics of interactions between pairs of chemical functional groups. Dr. Jason Cole is a Senior Research Fellow at CCDC.
"Using CSD data for this type of analysis provides different insights from energy-calculation-based methods, as the interactions observed in the CSD are influenced by more than their strength," says Cole.
Impacts of the study
Exscientia is a global leader in pharmatech, which sits at the interface of advanced AI application and complex drug discovery. They have implemented the hotspot mapping in-house within multiple drug discovery programs and use it to guide target validation and drug design. In addition, a research team at the University of Cambridge recently published in Nature how they used fragment hotspot mapping to identify structures that may assist in designing DNA-dependent protein kinase catalytic subunit inhibitors, which show potential as cancer therapeutics.
More information: Mihaela D. Smilova et al, Fragment Hotspot Mapping to Identify Selectivity-Determining Regions between Related Proteins, Journal of Chemical Information and Modeling (2022). DOI: 10.1021/acs.jcim.1c00823
Peter R. Curran et al, Hotspots API: A Python Package for the Detection of Small Molecule Binding Hotspots and Application to Structure-Based Drug Design, Journal of Chemical Information and Modeling (2020). DOI: 10.1021/acs.jcim.9b00996
Shikang Liang et al, Structural insights into inhibitor regulation of the DNA repair protein DNA-PKcs, Nature (2022). DOI: 10.1038/s41586-021-04274-9
Journal information: Journal of Chemical Information and Modeling , Nature
Provided by CCDC - Cambridge Crystallographic Data Centre