This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
AI approach optimizes development of antibody drugs

Proteins have evolved to excel at everything from contracting muscles to digesting food to recognizing viruses. To engineer better proteins, including antibodies, scientists often iteratively mutate the amino acids—the units that are arranged in a sequence to make up proteins—in different positions until the resulting protein has an improved function, like eliciting a stronger immune response or capturing carbon dioxide from the atmosphere more efficiently.
But there are more possible amino acid sequences than there are grains of sand in the world. And finding the best protein and, therefore, the best potential drug, is often expensive or impossible.
Stanford scientists have developed a new machine learning-based method to more quickly and accurately predict the molecular changes that will lead to better antibody drugs.
Published in Science on July 4, the approach combines the 3D structure of the protein backbone with large language models based on amino acid sequence, and allows researchers to find, in minutes, rare and desirable mutations that would otherwise only be found with exhaustive experiments.
Led by Peter S. Kim, professor of biochemistry and institute scholar at Sarafan ChEM-H, and Brian Hie, assistant professor of chemical engineering, the team showed that they could improve a once FDA-approved SARS-CoV-2 antibody that had been discontinued due to its ineffectiveness against a new strain in November 2022. Their approach resulted in a 25-fold improvement against the virus.
"A lot of effort in AI and drug development is centered around amassing tons of data about how well a certain molecule performs a certain task so that a computer can learn enough to design a better version," said Kim. "What's remarkable is that we've shown that structure can be used in lieu of a lot of that data, and the computer will still learn."
"Now, more antibodies actually get a shot at being optimized," said Hie, who is also an innovation investigator at the Arc Institute.
Bent into shape
When faced with the challenge of finding the best amino acid sequence, scientists will often make millions and test them in miniaturized, simplified versions of biological systems. They hope that the best drug in a dish will also be the best drug in humans.
"It's a lot of guess and check," said Hie. "The goal of a lot of intelligent algorithms is to remove the guesswork from this."
To speed up the process, scientists have developed ChatGPT-like machine learning algorithms that are trained on the amino acid sequences of millions of proteins to predict desirable mutations.
These models, however, often point scientists toward sequences that, once produced in the lab, are unstable or worse than where they started.
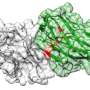
This is partially because protein function depends not only on the sequence of amino acids but also on the 3D structure of that sequence. For example, to trigger an immune response, antibodies must be the right shape to bind to molecules that sit atop the surface of viruses.
The key, the team thought, to developing a better prediction algorithm was structure. So, they constrained the long list of possible beneficial mutations—as determined by the sequence-based large language model—to only those that would preserve the 3D shape of the starting protein.
Testing ground
In December 2022, the team put it to the test on a recently discontinued SARS-CoV-2 antibody therapy.
"The prevailing theory was that trying to improve this antibody would fail," said Varun Shanker, a medical student, graduate student in biophysics, and lead author on the study. "The virus was too smart. It evolved as it spread through millions of people to know exactly how to mutate to avoid these antibodies."
Using purely sequence-based models to optimize the protein resulted in a modest two-fold increase in effectiveness. But with their structure-guided approach, the team saw a 25-fold increase.
"We were finally catching up to the virus," said Shanker, who is also a fellow in the Chemistry/Biology Interface Training Program at Sarafan ChEM-H.
Teaching an old model new tricks
Most efforts in using AI to build better drugs rely on "training" or "supervising" the model, which involves generating huge amounts of data about the function and performance of unique protein sequences. This approach takes a lot of time, and results in a model tailored to specific protein performing a specific task.
This model does not require any input about what the protein does, how well it does it, or any lab experiments. Because structure is so closely tied to function, the protein's coordinates become a proxy for performance.
For the COVID antibody work, they constrained the structure not just to the antibody itself, but to the antibody when it is bound to the virus. From there, their model "learned" some rules of antibody binding without ever needing to be taught.
Early experiments show that the approach is generalizable to other kinds of proteins, like enzymes, which help catalyze chemical reactions in our bodies. So far, the researchers have found that the model points scientists to tens of proteins, and, on average, half are better than the starting point.
This tool could be useful to quickly respond to emerging or evolving diseases. It also lowers the barrier to making more effective medicines.
Stronger medicines mean lower doses are necessary, which means that a given quantity could benefit more patients. For infectious diseases like HIV, where studies have shown that large but infrequent doses of an antibody can protect patients from infection, this could be transformational.
The team is making their model and code freely available to anyone.
"This is an exciting example of the power of deep learning to democratize the process of building better proteins," said Shanker. "This not only allows people to develop new medicines, but also opens up new areas of scientific exploration that had been inaccessible."
More information: Varun R. Shanker et al, Unsupervised evolution of protein and antibody complexes with a structure-informed language model, Science (2024). DOI: 10.1126/science.adk8946
Journal information: Science
Provided by Stanford University