Machine learning solves the who's who problem in NMR spectra of organic crystals

Solid-state nuclear magnetic resonance (NMR) spectroscopy—a technique that measures the frequencies emitted by the nuclei of some atoms exposed to radio waves in a strong magnetic field—can be used to determine chemical and 3D structures as well as the dynamics of molecules and materials.
A necessary initial step in the analysis is the so-called chemical shift assignment. This involves assigning each peak in the NMR spectrum to a given atom in the molecule or material under investigation. This can be a particularly complicated task. Assigning chemical shifts experimentally can be challenging and generally requires time-consuming multi-dimensional correlation experiments. Assignment by comparison to statistical analysis of experimental chemical shift databases would be an alternative solution, but there is no such database for molecular solids.
A team of researchers including EPFL professors Lyndon Emsley, head of the Laboratory of Magnetic Resonance, Michele Ceriotti, head of the Laboratory of Computational Science and Modeling and Ph.D. student Manuel Cordova decided to tackle this problem by developing a method of assigning NMR spectra of organic crystals probabilistically, directly from their 2D chemical structures.
They started by creating their own database of chemical shifts for organic solids by combining the Cambridge Structural Database (CSD), a database of more than 200,000 three-dimensional organic structures, with ShiftML, a machine learning algorithm they had developed together previously that allows for the prediction of chemical shifts directly from the structure of molecular solids.
Initially described in a Nature Communications paper in 2018, ShiftML uses DFT calculations for training, but can then perform accurate predictions on new structures without performing additional quantum calculations. Though DFT accuracy is attained, the method can calculate chemical shifts for structures with ~100 atoms in seconds, reducing the computational cost by a factor of as much as 10,000 compared to current DFT chemical shift calculations. The accuracy of the method does not depend on the size of the structure examined and the prediction time is linear in the number of atoms. This sets the stage for calculating chemical shifts in situations where it would have been unfeasible before.
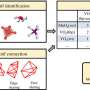
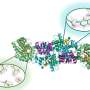
In the new Science Advances paper, the team used ShiftML to predict shifts on more than 200,000 compounds extracted from the CSD and then related the shifts obtained to topological representations of the molecular environments. This involved constructing a graph representing the covalent bonds between the atoms in the molecule, extending it a given number of bonds away from the central atoms. They then brought together all the identical instances of the graph in the database, allowing them to obtain statistical distributions of chemical shifts for each motif. The representation is a simplification of the covalent bonds around the atom in a molecule and doesn't contain any 3D structural features: this allowed them to obtain the probabilistic assignment of the NMR spectra of organic crystals directly from their two-dimensional chemical structures through a marginalization scheme that combined the distributions from all the atoms in the molecule.
After constructing the chemical shift database, the scientists looked to predict the assignments on a model system and applied the approach to a set of organic molecules for which the carbon chemical shift assignment has already, at least in part, been determined experimentally: theophylline, thymol, cocaine, strychnine, AZD5718, lisinopril, ritonavir and the K salt of penicillin G. The assignment probabilities obtained directly from the two-dimensional representation of the molecules were found to match the experimentally determined assignment in most cases.
Finally, they evaluated the performance of the framework on a benchmark set of 100 crystal structures with between 10 and 20 different carbon atoms. They used the ShiftML predicted shifts for each atom as the correct assignment and excluded them from the statistical distributions used to assign the molecules. The correct assignment was found among the two most probable assignments in more than 80% of cases.
"This method could significantly accelerate the study of materials by NMR by streamlining one of the essential first steps of these studies," Cordova said.
More information: Manuel Cordova et al, Bayesian probabilistic assignment of chemical shifts in organic solids, Science Advances (2021). DOI: 10.1126/sciadv.abk2341. www.science.org/doi/10.1126/sciadv.abk2341
Journal information: Science Advances , Nature Communications
Provided by Ecole Polytechnique Federale de Lausanne