May 10, 2021 feature
Structure motif-centric learning framework for inorganic crystalline systems
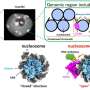
 and the generation of global motif representations using the motif environment matrix. Credit: Science Advances, doi: 10.1126/sciadv.abf1754")
Physical principles can be incorporated in a machine learning architecture as a fundamental setup to develop artificial intelligence for inorganic materials. In a new report now on Science Advances, Huta R. Banjade, and a research team in physics, computer and information science and nanoscience in the U.S. and Belgium proposed structure motifs in inorganic crystals to serve as a central input to a machine learning framework. The team demonstrated how the presence of structure motifs and their connections in a large set of crystalline compounds could be converted into unique vector representations via an unsupervised learning algorithm. They accomplished this by creating a motif-centric leaning framework by combining motif information with atom-based graph neural networks to form an atom-motif dual graph network (AMDNet). The setup accurately predicted the electronic structure of metal oxides such as bandgaps. The work illustrates a method to design graph neural network learning architectures to investigate complex materials beyond atom physical properties.
ML methods
Machine learning (ML) methods can be combined with massive material data to accelerate the discovery and rational design of functional solid-state compounds. Supervised learning can lead to material property predictions, including phase stability and crystal nature, effective for molecule dynamics simulations. Structure motifs can be created in accordance with Pauling's first rule, by forming a coordinated polyhedron of anions about each cation in a compound to behave as fundamental building blocks that are highly correlated with material properties. For instance, the structure motifs in crystalline compounds can play an essential role to determine the material properties in various technical and scientific applications. In this work, Banjade et al. incorporated structure motif information into a machine leaning (ML) framework. The scientists combined the motif information with graph convolutional neural networks to develop a motif-centric deep learning architecture known as atom-motif dual graph network (AMDNet). The accuracy of the structure surpassed that of an existing state-of-the-art atom-based graph network to predict the electronic structures of inorganic crystalline materials.
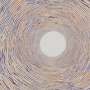
 cube, (2) cuboctahedron, (3) octahedron, and (4) a mixture of tetrahedron (in magenta) and square plane (in remnant). t-SNE, t-distributed stochastic neighbor embedding. Credit: Science Advances, doi: 10.1126/sciadv.abf1754")
An unsupervised learning algorithm Atom2Vec can understand high-dimensional vector representations of atoms by encoding basic properties of atoms based on an extensive database of chemical formulae. Banjade et al. focused on binary and ternary metal oxides that constitute a vast and diverse material space where crystal structures are characterized via cation-oxygen coordination. To extract the structure motif information, the team used the local environment identification method developed by Waroquiers et al. as implemented by the Pymatgen code. The team identified three different types of connectivity between a motif and its neighboring motif; including inner sharing (one-atom shared), edge sharing (two atoms shared), and face sharing (three or more atoms shared). The scientists then proposed a learning algorithm to take advantage of the motif data collection process and effectively converted each row of the motif environment matrix into a high-dimensional vector to represent a unique structure motif. They then extracted motif information for the learning process using a graph convolutional network. The team aimed to identify patterns and clustering information for these high-dimensional motif vectors to influence the complex material properties of oxide compounds. They visualized the high dimensional data using the t-distributed stochastic neighbor embedding (t-SNE) – a nonlinear dimensionality reduction technique.
Using motif information in graph neural networks.

The scientists obtained projected motif vector data in two dimensions using the t-SNE process. They noted distinct clusters based on the motif types. The chemical properties of the elements forming the motifs played a key role during cluster formation. For example, Lanthanide-based motifs formed different clusters on the basis of motif type and Yttrium-based motifs remained close to the Lanthanide-based motifs due to their chemical similarities. Motifs associated with zinc and magnesium also clustered together. The unsupervised learning-based findings supported the structure motifs to serve as essential inputs for crystalline compounds carrying elemental and structural information. The team then used structure motif information as an essential input to a graph neural network (GNN) to predict physical properties of materials. Most of the graph networks applied to crystalline materials. To enable a learning architecture of atom-level and motif-level graph representations of materials, Banjade et al. proposed that AMDNet could be constructed to enhance the learning process and improve the prediction accuracy for the electronic structure properties of metal oxides. In the motif graphs, the researchers encoded atom-level and motif-level information in each node and constructed the motif graph, including extended connectivity, angle, distance and order parameters using Python package robocrystallography.
AMDNet
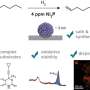
In the proposed AMDNet architecture, Banjade et al. incorporated motif information into a graph network learning framework to generate motif graphs and atom graphs representing compounds with different cardinality of edges and nodes to combine the information prior to making predictions. For each material, the team generated an atom graph and a motif graph. They then used 22,606 binary and ternary metal oxides from the Materials Project database to test the effectiveness of the proposed model and focused on the prediction of bandgaps—a complex electronic structure problem. The results showed the superiority of AMDNet during bandgap prediction when compared to preceding networks. The model also showed superior performance during a metal versus nonmetal classification task. The work showed the initial efforts to incorporate high-level material information in deep learning models for solid-state materials.
 Demonstration of the learning architecture of the proposed atom-motif dual graph network (AMDNet) for the effective learning of electronic structures and other material properties of inorganic crystalline materials. (B) Comparison of predicted and actual bandgaps [from density functional theory (DFT) calculations] and (C) comparison of predicted and actual formation energies (from DFT calculations) in the test dataset with 4515 compounds. Credit: Science Advances, doi: 10.1126/sciadv.abf1754")
Outlook
In this way, Huta R. Banjade and colleagues showed how structure motifs in crystal structures could be combined with unsupervised and supervised machine learning methods to improve the effective representation of solid-state material systems. For complex electronic structures, the team included the structure and motif connection information in to an AMDNet model to outperform existing networks and predict the electronic bandgaps and metal versus nonmetal classification tasks. This general learning framework can be used to predict other materials properties including mechanical and excited state properties across two-dimensional materials and metal-organic frameworks.
More information: Banjade H.R. et al. Structure motif–centric learning framework for inorganic crystalline systems, Science Advances, DOI: 10.1126/sciadv.abf1754
Curtarolo S. et al. Predicting Crystal Structures with Data Mining of Quantum Calculations., Physical Review Letters, doi.org/10.1103/PhysRevLett.91.135503
Dong Y. et al. Bandgap prediction by deep learning in configurationally hybridized graphene and boron nitride, Computational Materials, doi.org/10.1038/s41524-019-0165-4
Journal information: Science Advances , Physical Review Letters
Provided by Science X Network
© 2021 Science X Network