New tool could help researchers design better cancer vaccines

A computational model could improve the selection of tumor antigens for personalized cancer vaccines that are now in early-stage clinical trials.
Every cell in the human body is coated with fragments of proteins called antigens that tell the immune system what's inside the cell. Antigens presented on cells that are infected by foreign invaders or have become rogue cancers prompt an immune attack. Such antigens are often used in vaccines to spur immune responses against, for example, viruses like the flu. But to make vaccines that effectively stimulate attack against cancer, researchers need to predict exactly which tumor-specific antigens will be displayed on tumor cells and hence would be the best ones to put in a cancer vaccine.
Now, scientists at the Broad Institute of MIT and Harvard, Dana-Farber Cancer Institute, and Massachusetts General Hospital have developed a new computational tool that could help with this task. The researchers turned to machine learning to analyze a diverse set of more than 185,000 human antigens that they discovered, and generated a new set of rules that predict which antigens are presented on the surface of a person's cells. The findings, published today in Nature Biotechnology, could aid in the development of new treatments that stimulate the immune system to attack cancer as well as viruses and bacteria.
"Our goal is to be able to predict antigens on a personalized level with perfect accuracy," said Nir Hacohen, co-director of the Broad Institute's Center for Cell Circuits, director of the Center for Cancer Immunology at Massachusetts General Hospital, and a co-senior author of the study. "We're much closer to that goal now, but the field still has more to do."
Antigen diversity
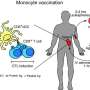
Figuring out a person's tumor antigens is tricky because they vary from person to person. A cellular program known as the antigen presentation system creates antigens by chopping up proteins inside any given cell and then using human leukocyte antigen (HLA) proteins to bind to and display a small subset of these fragments on the cell surface. But HLA genes are the most diverse human genes, resulting in more than 10,000 different "HLA types" across the human population. This translates to a large diversity in the protein fragments that end up on the cell surface.
Researchers can use simple blood tests to determine a person's HLA type. But cancers are so diverse between people that knowing someone's HLA type isn't enough to predict what's on the surface of tumor cells.
"If you can take data on a person and their cancer and be able to predict what tumor-specific antigens are displayed, that can help us induce an immune response against those antigens, through a vaccine or other mechanism," said Catherine Wu, an oncologist, chief of the Division of Stem Cell Transplantation and Cellular Therapies at Dana-Farber Cancer Institute, Institute Member at the Broad Institute, and co-senior author of the paper.
In the new work, Wu and Hacohen collaborated with Steven Carr, senior director of proteomics at the Broad, Derin Keskin of Dana-Farber Cancer Institute and others. The team isolated all HLA-associated protein fragments from 95 human cell lines that represented both common and rare HLA types in different populations. The researchers then used mass spectrometry to characterize these antigens. The resulting dataset included sequences of 186,464 protein fragments, or peptides. The team looked for relationships between the HLA types and the peptides, but realized they would need sophisticated tools to address this problem systematically.
"We explored various patterns in the data," said Sisi Sarkizova, a graduate student in the Hacohen lab and co-first author of the new paper. "But we needed to turn to machine learning to make better predictions of whether a new, unseen antigen would be presented or not."
Call in the computers
Using a machine learning approach, the research team inputted each of the antigen sequences, as well as the HLA type of the cell the antigens came from, into a computer program. The program parsed the data and determined new rules dictating which antigens are presented by each HLA type. Key factors that influenced which peptides were presented by cells included the length of the peptides, their expression levels, specific sequences that allow them to bind to HLA proteins, and other chemical properties.
The team also discovered that some peptides were displayed by more than one HLA type. "That was something we didn't expect, and could be really nice for vaccine development, because a single vaccine could potentially cover more people with the same antigens," said Keskin.
To test the effectiveness of the new rules, the team inputted a second set of data, from 11 human tumor samples—three chronic lymphocytic leukemia, one ovarian, three glioblastoma and four melanoma—into the model. "It identified nearly twice as many antigens than previous approaches, and correctly predicted more than 75 percent of the HLA-bound peptides that were detected using mass spectrometry," said Susan Klaeger, a postdoctoral fellow in Carr's group, and co-first author of the study.
The new model, which will be freely available for other researchers to use, could help researchers design not only better cancer vaccines, but also vaccines against pathogens such as human immunodeficiency virus (HIV) that mutate quickly and vary between people.
The researchers are working to further improve the accuracy of the model, and are also integrating it into ongoing clinical trials of cancer vaccines, to more effectively match vaccines to patients.
"We've already shown some evidence of being able to induce an anti-tumor immune response by predicting antigens on a personalized level," said Wu. Keskin added, "Our new methods will help us do that even better, and our analysis of 95 common and rare HLA alleles will make it feasible to predict tumor antigens for most populations world-wide."
More information: Siranush Sarkizova et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population, Nature Biotechnology (2019). DOI: 10.1038/s41587-019-0322-9
Journal information: Nature Biotechnology
Provided by Broad Institute of MIT and Harvard