Researchers develop machine learning tool to refine personalized immunotherapy

Ludwig Cancer Research scientists have developed a new and more accurate method to identify the molecular signs of cancer likely to be presented to helper T cells, which stimulate and orchestrate the immune response to tumors and infectious agents. The study, led by David Gfeller and Michal Bassani-Sternberg of the Lausanne Branch of the Ludwig Institute for Cancer Research, is reported in the current issue of Nature Biotechnology.
The new method combines two powerful new technologies. One is a mass spectrometry technology developed by Bassani-Sternberg's lab to rapidly and inexpensively obtain the amino acid sequences of thousands of peptide antigens—or protein fragments—bound to a molecular complex known as HLA that is expressed on the surface of cells. The other is a novel computational tool developed in Gfeller's lab that is based on machine learning, the computational approach that powers face-recognition software, among other things.
"This method advances our effort to find good targets for cancer immunotherapy," says Bassani-Sternberg. "But it is not only important for vaccines and other immunotherapies. It is also a tool we will be using for basic science, to better understand the interaction of cancers with the immune system."
Sick and cancerous cells chop up aberrant proteins associated with their disease and display fragments of those proteins on their surfaces to sound an immune alarm. They do so using two broad classes of HLA molecules: HLA-I, which activates the killer T cells that destroy cancerous or infected cells, and HLA-II, which activates helper T cells.
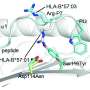
Distinct amino acid sequences—or binding "motifs"—direct the binding of antigenic peptides to each class of HLA molecules; those that lack such motifs are not presented by HLA molecules as targets of the immune response. Knowing those motifs thus allows researchers to select the right collection of peptides to serve as targets for (currently experimental) immunotherapies that are tailored to the unique molecular profile of an individual's cancer.
Researchers are already quite good at identifying peptides likely to bind HLA-I. But HLA-II has proved more problematic. "Binding to HLA-II is much less well understood," says Gfeller. "But we know from immunology that HLA-II antigen presentation to helper T cells is absolutely critical to priming the immune response." The results of recent cancer vaccine studies have, moreover, indicated that helper T cells are vitally important to the induction of effective anti-tumor immune responses.
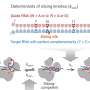
The rules of HLA-II binding have been hard to pin down due to the diversity of HLA-II molecules and the complexity of their peptide binding patterns. The researchers hypothesized that an unbiased analysis of amino acid patterns in the HLA-II bound peptides found by mass spectrometry might reveal some of those rules. "We believed it was very important to develop our own computational method because, that way, we could fine tune it to address this particular problem," says Gfeller.
To develop their predictive model, the Ludwig Lausanne team isolated more than 99,000 peptides bound to HLA-II molecules and determined their amino acid sequences. Gfeller and his postdoc Julien Racle fed this gargantuan dataset to their machine learning algorithm, MoDec (for motif deconvolution), and had it look for HLA-II binding motifs.
MoDec's results were used to train an algorithm to predict the HLA-II binding capability of peptides from a variety of tumors and pathogens. The predictor's identification of immunogenic HLA-II-binding peptides proved, by the researchers' reckoning, to be at least two-fold better than those achieved by previous efforts.
The predictor algorithm is already being implemented in Ludwig Lausanne's program to develop individualized immunotherapies for cancer. Further, the researchers point out that their work is likely to be useful to studies and interventions for autoimmune and infectious diseases as well.
More information: Julien Racle et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes, Nature Biotechnology (2019). DOI: 10.1038/s41587-019-0289-6
Journal information: Nature Biotechnology
Provided by Ludwig Institute for Cancer Research