Molecule studies reveal potential treatment for stroke patients

In an extension of research published a month ago in Nature Methods, a novel hybrid approach performed by researchers from Clemson University's department of physics and astronomy and Stony Brook University has revealed a 3-D structure of a protein fragment that could serve as a drug target in treating stroke patients.
The protein called "postsynaptic density protein of 95 kDa (PSD-95)" is positioned on neurons in the brain that are receiving chemical messages—neurotransmitters—from adjacent neurons. By recruiting receptors and other helper proteins, PSD-95 works to maintain the integrity of neural connections over time, thereby facilitating neural communication, learning and memory.
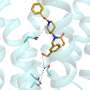
PSD-95 consists of five parts, or domains, that each play a different role in the protein's overall function. Two of these domains, called PDZ-1 and PDZ-2, have been shown to influence symptoms associated with ischemic stroke, such as paralysis or speech impairment.
"One of the ideas that has been postulated in the literature is to create a multivalent drug that targets both PDZ domains because they're very similar in nature. If you can block the PDZ domains from binding particular proteins or enzymes, you can reduce the debilitating effects of a stroke," said Hugo Sanabria, lead author on the study.
The challenge, however, is that it's nearly impossible to design a drug inhibitor without first knowing the exact structure of the PDZ domains of PSD-95. It would be like driving across the country without having a map of the United States.
"The biological functions of biomolecules are determined by their structures, so we need detailed structural and dynamic insights of PDZ-1 and -2 to help better understand their functional roles and aid in the design of novel inhibitors," said Feng Ding, Sanabria's colleague here at Clemson.
A handful of approaches exists to render the structure of biomolecules. But in the case of PSD-95, each approach—NMR spectroscopy, X-ray crystallography and Förster resonance energy transfer (FRET) - delivered a different structural model. The researchers' collaborator at Stony Brook University, associate professor Mark Bowen in the department of physiology and biophysics, established a partnership with Sanabria on this project after he uncovered one of the inconsistent structural models of the PSD-95 fragment.
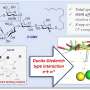
Sanabria's lab addressed this discrepancy by first modeling the PSD-95 fragment using FRET, an approach that identifies possible configurations of biomolecules. Under this method, Sanabria attached two light-sensitive molecules, called chromophores, at two differing positions on the PSD-95 fragment. He then uncovered the distance between the chromophores by visualizing the fragment under a microscope. This was repeated multiple times from different attaching points.
"For the modeling aspect, FRET gives you distances between chromophores, but that's not enough to fill all of the geometrical restraints of the molecule, so we have to rely on something else, some other methodology. That's where Professor Ding comes into play," Sanabria said.
Ding leads a computational biophysics lab at Clemson University where he uses computer software to gauge how biomolecules look, move and function. His approach to modeling utilizes a computer simulation known as discrete molecular dynamics (DMD) that maps the landscape of a biomolecule, predicting the trajectories of proteins as they fold and interact with other molecules. The subsequent simulation can be played back like a movie, helping researchers visualize protein behaviors over time.
"If you do traditional molecular simulations, typically you're going to sample a very tiny region of the space, particularly for larger molecules, so you're not going to have a good overview of how the entire molecule will look even in physiological conditions," Sanabria said. "Discrete molecular dynamics is a much faster and less computationally expensive way to accurately and rapidly sample the conformational space of proteins."
To do it, Sanabria first obtained a set of distances by measuring PSD-95 with FRET. In that experiment, Sanabria had 10 samples of the PSD-95 fragment that each were rendering different distances and three common shapes—or conformations—of PSD-95 were observed. Yet, without a DMD simulation, there was no way for the researchers to know which distance corresponded to which conformation of the fragment. So they input each possible distance against each possible shape and let the simulation do the rest.
"Once we did the first simulation, we saw that there were three main states that PDZ-1 and -2 were taking. One showed very close contact between the two, one showed a set of intermediate contact and one had no contact whatsoever," Ding said.
The researchers then ran a DMD simulation again without considering the FRET distances to confirm that the three observed states exist in nature and are not simply a fluke imposed by the FRET distances. They further probed the structures by looking at the way that individual amino acids, which constitute the PDZ domains, bond to one another. From these analyses, Ding, Bowen and Sanabria were able to confirm that the PDZ domains take on two out of the three observed states in the DMD simulation—that with some contact and that with no contact whatsoever.
"Now, we have two potential targets for engineering new drugs that will be more efficient than the ones that are currently available," Sanabria said. "The outlook for stroke patients is promising."
Without discrete molecular dynamics, which can capture conformational changes that occur on the microsecond timescale, these two states would have been missed as they were in past studies.
"Most of the people doing FRET-guided structural modeling are working with a rigid molecule, like DNA. If you have a rigid molecule, it's easy to model—you have only a single state to capture. You can assign the FRET distances and there's really no problem," Sanabria said. "In this case, we surpassed this approach in many ways."
In future studies, the team is looking to analyze the potential for the PSD-95 fragment to auto-inhibit itself based on the fragment's own structure.
More information: Inna S. Yanez Orozco et al, Identifying weak interdomain interactions that stabilize the supertertiary structure of the N-terminal tandem PDZ domains of PSD-95, Nature Communications (2018). DOI: 10.1038/s41467-018-06133-0
Björn Hellenkamp et al. Precision and accuracy of single-molecule FRET measurements—a multi-laboratory benchmark study, Nature Methods (2018). DOI: 10.1038/s41592-018-0085-0
Journal information: Nature Communications , Nature Methods
Provided by Clemson University