Researchers probe brain disease-causing proteins at the atomic level
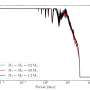
 between the following 13C atoms: A117Cβ-I139Cγ2, A117Cβ-I139Cδ, A117Cα-P137Cα, and G119Cα-S135Cβ. b Schematic model for the [hu] amyloid core based on the combination of solid-state NMR and tilted-beam transmission electron microscopy data (see text for details). In this model [hu] amyloid fibrils consist of two protofilaments in a C2-symmetric arrangement with β-sheet regions running parallel to the long fibril axis. The approximate locations of amino acid residues 112, 117, and 139, that have major impact on the structure adopted by PrP23-144 amyloid as discussed in the text, are indicated by red spheres. Credit: Nature Communications (2017). DOI: 10.1038/s41467-017-00794-z")
Researchers studying a protein that causes a hereditary degenerative brain disease in humans have discovered that the human, mouse and hamster forms of the protein, which have nearly identical amino acid sequences, exhibit distinct three-dimensional structures at the atomic level.
The protein causes familial human cerebral amyloid angiopathy (CAA), and the study, which appears in Nature Communications, is the first to examine forms of the protein in three different species.
Christopher Jaroniec, professor of chemistry and biochemistry at The Ohio State University, said that the findings highlight the fact that minor alterations in single amino acids can cause profound differences in structure and function among this family of proteins.
"The large-scale differences in the structures and transmission characteristics of these proteins—caused by what amounts to seemingly insignificant differences in the positions of a few carbon and hydrogen atoms—are quite remarkable," Jaroniec said.
The study doesn't form the basis for a new test or treatment for CAA, but rather uses these proteins as models for understanding the fundamental aspects of the cross-species transmission of a whole class of degenerative brain diseases known as prion diseases, he explained. It also underscores the utility of solid-state nuclear magnetic resonance (NMR) spectroscopy for imaging the structures of proteins associated with prion diseases.
Researchers know that in the body, the protein molecules associated with CAA form plaques that lodge in blood vessel walls in the brain, but there haven't been detailed examinations of the molecular structure of these plaques until recently. In 2008, Ohio State researchers and their partners at Case Western Reserve University performed the initial solid-state studies of the relevant prion protein variant, and narrowed the list of possibly critical amino acids for its function to about 30.
Now, they've demonstrated that a single amino acid—known by its number along the protein chain, 139—is the key to this prion protein variant adopting a "human-like" versus a "hamster-like" structure, while another amino acid, 112, governs the structural differences between the human and mouse versions of the protein. They have also shown that these two amino acids appear to be responsible for the emergence of structurally distinct "prion strains" within the same protein sequence, in analogy to distinct strains of a virus.
The most well-known prion diseases include bovine spongiform encephalopathy (often called "mad cow disease") and Creutzfeldt-Jakob disease in humans. All are incurable and fatal, and some can also be transmissible. The structures adopted by the brain prion proteins within the plaques are thought to be critical to their ability to be transmitted between different hosts and cause disease.
"Our group is currently working on determining the high resolution molecular structures of the truncated prion protein variants associated with familial human CAA in order to gain a complete atomistic understanding of the factors underlying their transmission, and the present study is a major stepping stone in this effort," Jaroniec said.
"We hope that one day our group and other researchers will be able to use similar methodologies to unravel the structural basis of the transmissible prion diseases," he added.
More information: Theint Theint et al. Species-dependent structural polymorphism of Y145Stop prion protein amyloid revealed by solid-state NMR spectroscopy, Nature Communications (2017). DOI: 10.1038/s41467-017-00794-z
Journal information: Nature Communications
Provided by The Ohio State University