Benchmarking computational methods for metagenomes

They are everywhere, but invisible to the naked eye. Microbes are the unseen, influential forces behind the regulation of key environmental processes such as the carbon cycle, yet most of them remain unknown. For more than a decade, the U.S. Department of Energy Joint Genome Institute (DOE JGI), a DOE Office of Science User Facility, has been enabling researchers to study uncultured microbes unable to grow in the lab, using state-of-the-art approaches such as high-throughput genomic sequencing of environmental communities ("metagenomics") and the development of computational tools to uncover and characterize microbial communities from the environment. To tackle assembling metagenomes into a set of overlapping DNA segments that together represent a consensus region of DNA or contigs, then binning these contigs into genome bins, and finally conducting taxonomic profiling of genome bins, analysts around the world have developed an array of different computational tools, however until now there was a lack of consensus on how to evaluate their performance.
Published October 2, 2017 in Nature Methods, a team including DOE JGI researchers described the results of the Critical Assessment of Metagenome Interpretation (CAMI) Challenge, the first-ever, community-organized benchmarking assessment of computational tools for metagenomes. The CAMI Challenge was led by Alexander Sczyrba, head of the Computational Metagenomics group at Bielefeld University and formerly a DOE JGI postdoctoral fellow, and Alice McHardy, head of the Computational Biology of Infection Research Lab at the Helmholtz Centre for Infection Research.
"It is very difficult for researchers to find out which program to use for a particular data set and analysis based on the results from method papers," said McHardy. "The data sets and evaluation measures used in evaluations vary widely. Another issue is that developers usually spend a lot of time benchmarking the state-of-the-art when assessing the performance of novel software that way. CAMI wants to change these things and involves the community in defining standards and best practices for evaluation and to apply these principles in benchmarking challenges."
The CAMI Challenge took place over three months in 2015. To assess the computational tools, the organizers developed 3 simulated metagenome datasets using more than 300 draft genomes of bacterial and archaeal isolates sequenced and assembled by the DOE JGI, which were part of the Genomic Encyclopedia of Bacteria and Archaeal project published recently in Nature Biotechnology. These genomes were shared with to the CAMI Challenges consortium before being released to the public to facilitate the objective benchmarking of different tools. The datasets also included around the same number of genomes from the Max Planck Institute in Cologne, Germany, along with circular elements and viruses. The simulated datasets were a single sample dataset of 15 billion bases (Gb), a 40 Gb dataset with 40 genomes and 20 circular elements, and a 75 Gb time series data set comprised of five samples and including hundreds of genomes and circular elements.
"JGI has a strong interest in benchmarking of tools and technologies that would advance the analysis of metagenomes and improve the quality of data we provide to the users. Having published the very first study on the use of simulated datasets for benchmarking of metagenomics tools from the JGI, it is great to see how this methodology has expanded over the years and now through this study, evolving into a model for standardized community efforts in the field," said Nikos Kyrpides, DOE JGI Prokaryote Super Program head.
"JGI is very vested in not only benchmarking of lab protocols, but also computational workflows," added DOE JGI Microbial Genomics head Tanja Woyke. "This makes our participation in critical community efforts such as CAMI so important."
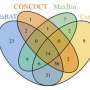
With more than 40 teams signed up for the Challenge, and the CAMI organizers received 215 submissions from 25 programs around the world, though only 17 teams were willing to have their software implementations published. The CAMI organizers evaluated computational tools in 3 categories. Half a dozen assemblers and assembly pipelines were evaluated on assembling genome sequences generated from short-read sequencing technologies. In the binning challenge, five genome binners and 4 taxonomic binners were evaluated on criteria including the tools' efficacy in recovering individual genomes. Finally, 10 taxonomic profilers with various parameter settings were evaluated on how well they could predict the identities and relative abundances of the microbes and circular elements. The benchmarking results are available on https://data.cami-challenge.org/results.
The CAMI organizers are already planning future benchmarking challenges, for example to evaluate and aid method development for long read sequencing technologies. "CAMI is an ongoing initiative," noted Sczyrba. "We are currently further automating the benchmarking and comparative result visualizations. And we invite everyone interested to join and work with CAMI on providing comprehensive performance overviews of the computational metagenomics toolkit, to inform developers about current challenges in computational metagenomics and applied scientists of the most suitable software for their research questions."
More information: Alexander Sczyrba et al, Critical Assessment of Metagenome Interpretation—a benchmark of metagenomics software, Nature Methods (2017). DOI: 10.1038/nmeth.4458
Journal information: Nature Methods , Nature Biotechnology
Provided by DOE/Joint Genome Institute