International competition benchmarks metagenomics software

Communities of bacteria live everywhere: inside our bodies, on our bodies and all around us. The human gut alone contains hundreds of species of bacteria that help digest food and provide nutrients, but can also make us sick. To learn more about these groups of bacteria and how they impact our lives, scientists need to study them. But this task poses challenges, because taking the bacteria into the laboratory is either impossible or would disrupt the biological processes the scientists wish to study.
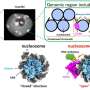
To bypass these difficulties, scientists have turned to the field of metagenomics. In metagenomics, researchers use algorithms to piece together DNA from an environmental sample to determine the type and role of bacteria present. Unlike established fields such as chemistry, where researchers evaluate their results against a set of known standards, metagenomics is a relatively young field that lacks such benchmarks.
Mihai Pop, a professor of computer science at the University of Maryland with a joint appointment in the University of Maryland Institute for Advanced Computer Studies, recently helped judge an international challenge called the Critical Assessment of Metagenome Interpretation (CAMI), which benchmarked metagenomics software. The results were published in the journal Nature Methods on October 2, 2017.
"There's no one algorithm that we can say is the best at everything," said Pop, who is also co-director of the Center for Health-related Informatics and Bioimaging at UMD. "What we found was that one tool does better in one context, but another does better in another context. It is important for researchers to know that they need to choose software based on the specific questions they are trying to answer."
The study's results were not surprising to Pop, because of the many challenges metagenomics software developers face. First, DNA analysis is challenging in metagenomics because the recovered DNA often comes from the field, not a tightly controlled laboratory environment. In addition, DNA from many organisms—some of which may not have known genomes—mingle together in a sample, making it difficult to correctly assemble, or piece together, individual genomes. Moreover, DNA degrades in harsh environments.
"I like to think of metagenomics as a new type of microscope," Pop said. "In the old days, you would use a microscope to study bacteria. Now we have a much more powerful microscope, which is DNA sequencing coupled with advanced algorithms. Metagenomics holds the promise of helping us understand what bacteria do in the world. But first we need to tune that microscope."
CAMI's leader invited Pop to help evaluate the submissions by challenge participants because of his expertise in genome and metagenome assembly. In 2009, Pop helped publish Bowtie, one of the most commonly used software packages for assembling genomes. More recently, he collaborated with the University of Maryland School of Medicine to analyze hundreds of thousands of gene sequences as part of the largest, most comprehensive study of childhood diarrheal diseases ever conducted in developing countries.
"We uncovered new, unknown bacteria that cause diarrheal diseases, and we also found interactions between bacteria that might worsen or improve illness," Pop said. "I feel that's one of the most impactful projects I've done using metagenomics."
For the competition, CAMI researchers combined approximately 700 microbial genomes and 600 viral genomes with other DNA sources and simulated how such a collection of DNA might appear in the field. The participants' task was to reconstruct and analyze the genomes of the simulated DNA pool.
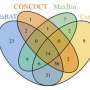
CAMI researchers scored the participants' submissions in three areas: how well they assembled the fragmented genomes; how well they "binned," or organized, DNA fragments into related groups to determine the families of organisms in the mixture; and how well they "profiled," or reconstructed, the identity and relative abundance of the organisms present in the mixture. Pop contributed metrics and software for evaluating the submitted assembled genomes.
Nineteen teams submitted 215 entries using six genome assemblers, nine binners and 10 profilers to tackle this challenge.
The results showed that for assembly, algorithms that pieced together a genome using different lengths of smaller DNA fragments outperformed those that used DNA fragments of a fixed length. However, no assemblers did well at picking apart different, yet similar genomes.
For the binning task, the researchers found tradeoffs in how accurately the software programs identified the group to which a particular DNA fragment belonged, versus how many DNA fragments the software assigned to any groups. This result suggests that researchers need to choose their binning software based on whether accuracy or coverage is more important. In addition, the performance of all binning algorithms decreased when samples included multiple related genomes.
In profiling, software either recovered the relative abundance of bacteria in the sample better or detected organisms better, even at very low quantities. However, the latter algorithms identified the wrong organism more often.
Going forward, Pop said the CAMI group will continue to run new challenges with different data sets and new evaluations aimed at more specific aspects of software performance. Pop is excited to see scientists use the benchmarks to address research questions in the laboratory and the clinic.
"The field of metagenomics needs standards to ensure that results are correct, well validated and follow best practices," Pop said. "For instance, if a doctor is going to stage an intervention based on results from metagenomic software, it's essential that those results be correct. Our work provides a roadmap for choosing appropriate software."
More information: Alexander Sczyrba et al, Critical Assessment of Metagenome Interpretation − a benchmark of computational metagenomics software, Nature Methods (2017). DOI: 10.1101/099127
Journal information: Nature Methods
Provided by University of Maryland