Scientists develop robust method for analysis of intestinal bacteria

A group of Russian scientists, among them staff at the Moscow Institute of Physics and Technology, have proposed a new method for the comparison of metagenome-coupled DNA sequences from all of the organisms in a sample of biological material being investigated. The method makes it possible to more effectively and quickly solve the task of comparing samples and can be easily embedded in the data-analysis process of any metagenome study. The study has been published in the BMC Bioinformatics journal.
The bacteria, which inhabit the human body, hold a special place for scientists in the study of metagenomics. The significance of metagenomics cannot be underestimated: Bacterial cells in our body outnumber our own by an order of magnitude and most of them are located in the gut. Global projects, such as the Human Microbiome Project, have revealed that the composition of the bacterial community affects our risk of disease, the selection of an optimal diet, mood and even creativity. The reverse is true – the composition of these microorganisms is sensitive to processes occurring in the body. Thus, by comparing the sample patient with people who have healthy intestinal metagenomes, it will be possible in the long term to evaluate the risk of dangerous diseases such as diabetes or inflammatory bowel disease.
The traditional approach to metagenome analysis is to compare samples on the basis of their taxonomic composition, percentages for each microbial species found. To determine the composition of the sample, its genetic sequences are compared with a database of known bacterial genomes called the reference set. However, this approach has several disadvantages. Firstly, the reference genomes are often inaccurate, since determining the composition of the reference genome is a computationally complex and time-consuming task, especially for species that are difficult to cultivate; and the genomes of species isolated in the laboratory can carry a set of genes significantly different from the same species living in a natural environment. Secondly, not all organisms are collected in reference genomes generally; examples of such organisms are viruses. Therefore, that part of the sample sequence that does not match with the reference sample is simply not taken into account during the analysis, despite the fact that it can be quite large and significant. Meanwhile, a method based on a comparison of k-mer frequencies does not require recourse to a reference sample or the existence of any information on the organisms studied; therefore, all sequences in the sample are subjected to analysis, which gives the best results.

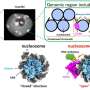
The method is based on the representation of an organism's genomic sequence as the set with all instances of nucleotide "words" of specified length "k", called k-mers. Because the genome is a unique sequence for each organism, the sets of such "words" also differ between individual organisms. Thus, the set of all k-mers for a metagenome can be viewed as a set of sets, namely of its constituent organisms. This allows assessment of the differences in the bacterial composition when comparing samples.
To test the effectiveness of the k-mer technique compared to traditional approaches, two sets of metagenome data were used – a set of real data and a set of artificially generated data. Artificial data (created from genomes, with proportions known beforehand) is convenient to use for testing the method, the sequence is precisely known and the results can be assessed by comparing them with an a priori correct value. Intestinal metagenomes from residents of the United States and China were used as real data.
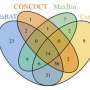
It is known that bacterial intestinal communities differ significantly between populations, and algorithms revea; exactly those indicators that show the difference in composition. Therefore, the criterion for assessing the effectiveness of the method was the extent to which the metagenomes can be distinguished—that is, how much the Chinese metagenomes differ in general from American ones.
The method has shown better results in both data types by comparing k-mers than when using traditional mapping with a reference set. In addition, when using real data, a mismatch between the intestinal results for k-mer and traditional approaches allowed the researchers to detect another important component of the intestinal metagenome—namely, the bacterial phage crAssphage, which had escaped the notice of researchers using the traditional method. The author of the article, Dmitri Alexeev, says, "Interestingly, the genes can be viewed not only as segments of DNA with proteins encoded in them, but also as information in general. It is this information distinction that has allowed us to identify new segments of DNA not described in the catalog of known genes. It is interesting to see how this approach will be used by other research groups."
The technique allows researchers to find the differences between the metagenomes for a variety of bacterial communities more efficiently and accurately, which can help to study, diagnose and treat many human diseases.
More information: Veronika B. Dubinkina et al. Assessment of k-mer spectrum applicability for metagenomic dissimilarity analysis, BMC Bioinformatics (2016). DOI: 10.1186/s12859-015-0875-7
Provided by Moscow Institute of Physics and Technology