Scientists use light to uncover the cause of sickle cell disease
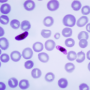
In sickle cell disease, hemoglobin—the oxygen-carrying component of blood—forms fibers that stiffen red blood cells and cause life-threatening symptoms. Using light-scattering techniques to study the detailed thermodynamics of this process, researchers reporting in the November 5 issue of the Biophysical Journal, a Cell Press publication, have determined the strength of the forces that hold these fibers intact. The information could be used to design therapies that interfere with the sickling process.
Red blood cells resemble beanbags whose contents are molecules of hemoglobin that give the cells their "squishiness" by slipping past one another rather than sticking together. This is no easy feat, because the molecules have positive and negative charges spread around their surfaces, as well as oily patches that repel water and thus provide natural partners. Yet it's thought that when two places of mutual attraction meet, locations of repulsion also come together and keep the net sum of attraction at zero.
In rare cases, such as sickle cell disease, a mutation in hemoglobin disrupts this delicate cancellation of attractive spots, and large stiff fibers, or polymers, of hemoglobin form inside normally pliable red blood cells.
Dr. Frank Ferrone, of Drexel University in Philadelphia, and Dr. Yihua Wang, currently at the Mayo Clinic in Rochester, discovered that the cancellation is not as perfect as previously thought, however. In fact, hemoglobin molecules do indeed associate, especially when the temperature is high or when hemoglobin solutions are concentrated. "This is true for normal hemoglobin, and even more pronounced for sickle hemoglobin," says Dr. Ferrone. His team found that under physiological conditions, sickle cell hemoglobin molecules are more likely to be found in pairs than as solitary molecules.
The researchers made these discoveries while conducting experiments with light-scattering techniques: they measured how rays of light are deflected by sickle hemoglobin fibers in order to calibrate the strength of the connections that hold them together. "By comparing the propensity of molecules of sickle hemoglobin to associate into pairs with the propensity of normal hemoglobin to do so, the relative strengths of the two major bonds within the sickle polymer were determined. The sickle cell mutation site was far stronger," Dr. Ferrone explains. "This makes the sickle hemoglobin polymers behave much like long tiny coil springs and helps us to understand their stiffness, which causes so much difficulty for affected individuals."
The findings could lead to new drug therapies that target the regions of hemoglobin that are responsible for polymer formation.
More information: Biophysical Journal, Wang et al.: "Dissecting the Energies that Stabilize Sickle Hemoglobin Polymers." dx.doi.org/10.1016/j.bpj.2013.09.032
Journal information: Biophysical Journal
Provided by Cell Press