This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
GPCR structure: Research reveals molecular origins of function for a key drug target

Through an international collaboration, scientists at St. Jude Children's Research Hospital have leveraged data science, pharmacology and structural information to conduct an atomic-level investigation into how each amino acid in the receptor that binds adrenaline contributes to receptor activity in the presence of this natural ligand.
They had discovered precisely which amino acids control the key pharmacological properties of the ligand. The adrenaline receptor studied is a member of the G protein-coupled receptor (GPCR) family, and this family is the target of one-third of all Food and Drug Administration (FDA)-approved drugs. Thus, understanding how GPCRs respond to natural or therapeutic ligands is critical for developing new therapies with precise effects on receptor activity.
The paper is published in the journal Science.
To understand how a watch works, one might take it apart, piece by piece, and study the role played by each component in its timekeeping function. Similarly, in a protein such as a GPCR, each amino acid might play a different role in how the protein responds to an external signal.
Researchers at St. Jude, in collaboration with scientists from Stanford University, the University of Montreal, the MRC Laboratory of Molecular Biology and Cambridge University, investigated the β2-adrenergic receptor (β2AR) by substituting one amino acid at a time to understand the contribution of each amino acid in this receptor to mediate a signaling response.
"Scientists learn how genes contribute to cell function by disrupting them one at a time. We asked, 'Why don't we take this one level deeper? Let's understand how every amino acid contributes to the functioning of a receptor by mutating them, one amino acid at a time,'" said co-corresponding author M. Madan Babu, Ph.D., from St. Jude's Department of Structural Biology, Center of Excellence for Data-Driven Discovery director and the George J. Pedersen Endowed Chair in Biological Data Science.
"Through evolution, every amino acid in the receptor has been sculpted in some way or another to ensure that it binds the natural ligand, in this case adrenaline, and elicits the appropriate physiological response."
Finding function in the form
GPCRs are proteins that span the cell's membrane and connect the outside of the cell to its internal environment by transmitting external signals to the inside of the cell. In the case of the β2AR, adrenaline binds to the GPCR on the part outside of the cell, inducing a response inside the cell.
When a ligand binds, it causes changes in the shape of the receptor, especially in the intracellular region of the receptor where a G protein binds. The binding sites for the ligand and the G protein are on opposite sides of the protein but connect through a complex network of amino acid contacts that span the entire protein. Conformational (shape) changes within the GPCR activate the G protein to trigger a downstream signaling response within the cell. Through effects on multiple tissues and GPCRs, including the β2AR, adrenaline can trigger the fight-or-flight response, such as during an adrenaline surge.
To understand the role of each amino acid in a GPCR, Franziska Heydenreich, Ph.D., from the Philipps University of Marburg, the lead and co-corresponding author of this project, mutated each of the 412 amino acids in the β2AR. She then evaluated each mutant's response to the ligand adrenaline and determined the classical pharmacological properties of efficacy and potency. Efficacy measures the maximum response a ligand can elicit, and potency measures the amount of ligand required to elicit half of the maximum response. The aim was to reveal, on an atomic scale, how each amino acid contributes to these pharmacological properties.
"Surprisingly, only about 80 of the more than 400 amino acids contributed to these pharmacological properties. Of these pharmacologically relevant amino acids, only one-third were located within regions where the ligand or G protein bound to the receptor," Heydenreich said.
"It was fascinating to observe that there are some amino acids that control efficacy, some that control potency and then there are others that affect both," Babu said. "It means if you want to make a more potent or efficacious drug, you now know there are specific residues that the new ligand needs to influence." The researchers also noted that the individual contribution of each residue to efficacy and potency was not equal, implying even more opportunities for fine-tuning drug responses while designing new therapeutic ligands.
"Efficacy and potency have been measured for numerous ligand-receptor signaling systems for several decades. Now we can understand how specific amino acids in a protein's sequence can influence these pharmacological properties," Babu explained.
"A fascinating aspect of the results is that potency and efficacy can be regulated independently of each other through distinct mechanisms. This provides a basis for understanding how genetic variation influences drug responses among individuals," Michel Bouvier, Ph.D., co-corresponding author from the Department of Biochemistry and Molecular Medicine and General Director of the Institute for Research in Immunology and Cancer of the University of Montreal added.
A beautiful network
Prior research illustrated the structure of both the active and inactive states of the β2AR. Building on this knowledge, the researchers embarked on a new investigation. They explored whether the two-thirds of pharmacologically relevant amino acids previously demonstrated to be not involved in ligand or G-protein binding might play a role in the transition between the active and inactive states of the receptor.
"We systematically started looking at every residue contact unique to the active state," Heydenreich said, "to understand whether all the amino acids that make an active-state contact are important."
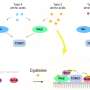
The researchers developed a data science framework to integrate pharmacological and structural data systematically and revealed the first comprehensive picture of GPCR signaling. "When we mapped the pharmacological data onto the structure, they formed a beautiful network," said Babu.
"It provided new insights into the allosteric network linking the ligand binding pocket to the G protein binding site that governs efficacy and potency," added Brian Kobilka, co-corresponding author and the 2012 Nobel Prize winner in Chemistry from Stanford University School of Medicine.
By understanding GPCR signaling at the atomic level, the researchers are optimistic that they can begin probing even deeper—to see the transient sub-states between the active and inactive conformations and to explore the conformational landscape of proteins.
"We now know which mutants to go after, those that only affect efficacy, potency or both," Heydenreich said.
"Now, we can perform molecular dynamics calculations and single-molecule experiments on those mutants to reveal the exact mechanisms by which the allosteric network influences efficacy and potency to mediate a signaling response. This is a direction we are pursuing through a St. Jude Research Collaborative on GPCRs that includes PIs from several institutions." Babu explained.
Apart from these "driver" residues that are involved in mediating active state-specific contacts and affect pharmacology when mutated, Babu and his colleagues intend to probe other key findings revealed by this work. They aim to study "passenger" amino acids that—despite making contacts in the active state—do not affect efficacy or potency when mutated.
They are also interested in "modulator" residues that don't mediate active state-specific contacts, but alter pharmacology when mutated. Their data science approach, integrating structural information and pharmacological measurements, isn't limited to the β2AR. It can be extended to any GPCR to enhance our understanding of the mechanics governing this crucial class of drug targets.
More information: Franziska M. Heydenreich et al, Molecular determinants of ligand efficacy and potency in GPCR signaling, Science (2023). DOI: 10.1126/science.adh1859. www.science.org/doi/10.1126/science.adh1859
Journal information: Science
Provided by St. Jude Children's Research Hospital