This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Alternative method cuts time for computer simulation of absorption spectrum from days to hour

Absorption spectroscopy is an analytical chemistry tool that can determine if a particular substance is present in a sample by measuring the intensity of the light absorbed as a function of wavelength. Measuring the absorbance of an atom or molecule can provide important information about electronic structure, quantum state, sample concentration, phase changes or composition changes, among other variables, including interaction with other molecules and possible technological applications.
Molecules with a high probability of simultaneously absorbing two photons of low-energy light have a wide array of applications: in molecular probes for high-resolution microscopy, as a substrate for data storage in dense three-dimensional structures, or as vectors in medicinal treatments, for example.
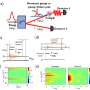
Studying the phenomenon by means of direct experimentation is difficult, however, and computer simulation usually complements spectroscopic characterization. Simulation also provides a microscopic view that is hard to obtain in experiments. The problem is that simulations involving relatively large molecules require several days of processing by supercomputers or months by conventional computers.
To overcome this difficulty, an alternative method of calculation has been proposed by physicist Tárcius Nascimento Ramos and collaborators in an article published in The Journal of Chemical Physics.
"We evaluated the performance of a semi-empirical method much used in past decades but more recently neglected by the scientific community owing to its approximative nature. Using this method, we were able to reduce calculation time to four hours in a conventional computer. The low computing cost enabled us to consider a large statistical sample for simulations of molecules in solutions, which isn't feasible with the currently hegemonic method," Ramos told Agência FAPESP.
The currently hegemonic method is density functional theory (DFT), a mathematical tool widely used in quantum mechanics to describe the electronic properties of complex systems without having to investigate the individual wave functions of each electron.
"The alternative method we used was INDO/S [intermediate neglect of differential overlap with spectroscopic parameterization]. It's based on the wave function of the molecular system but resolves approximately. Parts of the complex and computationally costly calculations are replaced by tabulated values obtained by adjusting experimental spectroscopic data. This makes the method highly efficient for theoretical studies of large molecular compounds," Ramos explained.
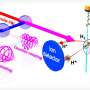
The practicality of this method can be sensed by taking into consideration the fact that the molecule studied, which is derived from stilbene, has more than 200 atoms (of carbon, oxygen and hydrogen). Besides the number of components, which alone would make conventional simulations extremely laborious and expensive, these large molecules have an additional complication: they are flexible, and their electronic properties change when they change shape (by twisting, for example).
"At the end of the study, we bridged the experimental gap by characterizing at the microscopic level the one- and two-photon absorption spectra for this class of molecules. We found that the semi-empirical method we tested, often neglected owing to its approximative nature, is the most suitable for predicting the one- and two-photon absorption spectra of large molecules in solution. This finding points to a route for molecular engineers to develop novel compounds with greater efficiency in their various application branches," Ramos said.
Here it may be useful to examine the difference between one- and two-photon absorption. The general principle is that molecules absorb photons only when they can assume excited states that are compatible with the energy of the photons.
The selection rules for one-photon absorption are not the same as for two-photon absorption, so that excited states prohibited for the former may be permitted for the latter. Owing to this difference, plus the high spatial resolution of excitation by two photons resulting from its non-linear optical nature, molecules that can absorb two photons are suitable for much more refined uses.
"Microscope imaging with two-photon absorption has far higher resolution and can be used to characterize deep tissue with less damage to the surrounding structures. In the case of data storage, the high resolution means 3D structures can be created with precision and plenty of detail, so that points inside materials can be encoded with high data density per volume," Ramos explained.
Computer modeling of two-photon absorption by organic molecules in solution was the subject of Ramos's Ph.D. research. The JCP article refers to another step forward in this investigation.
More information: Tárcius N. Ramos et al, Calculation of the one- and two-photon absorption spectra of water-soluble stilbene derivatives using a multiscale QM/MM approach, The Journal of Chemical Physics (2023). DOI: 10.1063/5.0152308
Journal information: Journal of Chemical Physics
Provided by FAPESP