Researchers adapt social network analysis to model virus evolution

Researchers from Western University may have discovered a new meaning to the social media phrase, "going viral."
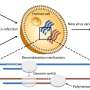
New research from Western University suggests some viruses evolve more like a dynamic social network—rather than a rigid tree, as was previously believed—recombining with one another to create a web of intersecting subtypes. This work has implications for epidemiology and public health, not just for HIV-1 but for other viruses as well, because it influences how we track the way viruses move through the population.
Previously, when epidemiologists look at the evolution of viruses, they classify variants like a growing tree—each variant with one or more mutations from its parent branching out into new variants.
By studying full-length genomes of HIV-1 from around the world, especially regions where sequencing has been less common, a team at Western's Schulich School of Medicine & Dentistry led by Art Poon, demonstrated that recombination is much more prevalent than previously believed.
"If people get infected with two or more subtypes of HIV, they exchange parts of their genomes to create mosaic forms that we call 'recombinants,'" said Abayomi Olabode, lead author and post-doctoral associate in Poon's lab at Western. "With our research and approach, we're finding that this recombination is not just a curiosity but happens much more than we originally thought."
The study, "Revisiting the recombinant history of HIV-1 group M with dynamic network community detection," was published this week in PNAS.
Typically, viruses like HIV-1 are classified by comparing only part of the virus' genome to a reference genome. However, Poon and his team developed a new computational method to look instead at the entire genome and discovered much more diversity within the subtypes. This finding shows there is extensive recombination throughout the evolution of HIV-1, including evidence that even the reference genomes of the virus may have been recombinants.
"This discovery was possible because we have more full-length genome data available now than there used to be, and there are more global initiatives to sequence populations that had been neglected in the past," said Poon, associate professor in pathology and laboratory medicine at Schulich Medicine & Dentistry.
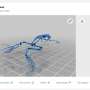
With this data in hand, the team was inspired by social network analysis to look at the data in a new way. When researchers analyze data from social networks like Facebook or Twitter, they have to take into account the intersecting and shifting links between people. "The analogy that we're making is that similar to dynamic social network analysis, as we go across the genome, parts of the genome will become more similar to a completely different group of other genomes because of recombination."
The team says this new approach has implications for how viruses are classified, and for tracking and understanding how viruses move through the population.
"Currently, if we want to know how an epidemic is spreading through a population, we compare the sequences, we build a tree, and then we use that tree to try to determine what's going on; where is the virus spreading faster than in other parts of the population, for example," said Poon. "What we've found is that recombination breaks those methods—so now we have to work on the assumption that there isn't just one tree, there is a whole forest depending on what part of the virus' genome you are looking at."
More information: Abayomi S. Olabode et al, Revisiting the recombinant history of HIV-1 group M with dynamic network community detection, Proceedings of the National Academy of Sciences (2022). DOI: 10.1073/pnas.2108815119
Journal information: Proceedings of the National Academy of Sciences
Provided by University of Western Ontario