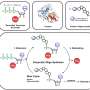
A molecular framework to bridge experimental and computer sciences for peptide-based materials engineering

Researchers in the Stephenson School of Biomedical Engineering, Gallogly College of Engineering, at the University of Oklahoma have developed a framework published in Science Advances that solves the challenge of bridging experimental and computer sciences to better predict peptide structures. Peptide-based materials have been used in energy, security and health fields for the past two decades.
Handan Acar, Ph.D., the Peggy and Charles Stephenson Assistant Professor of Biomedical Engineering at OU, teamed up with Andrew White, Ph.D., an associate professor of chemical engineering at the University of Rochester, to introduce a new strategy to study fundamentals of molecular engineering. Seren Hamsici, a doctoral student in Acar's lab, is the first author of the study.
Proteins are responsible for the structure, function and regulation of the body's organs and tissues. They are formed by amino acids and come together in different interactions, called intermolecular interactions, that are essential to how proteins perform different roles in the body. When these protein interactions behave abnormally, medical issues result, such as when they clump together to form plaques in the brain that leads to Alzheimer's Disease.
"In the peptide-engineering field, the general approach is to take those natural proteins and make incremental changes to identify the properties of the end aggregated products, and then find an application for which the identified properties would be useful," Acar said. "However, there are more than 500 natural and unnatural amino acids. Especially when you consider the size of the peptides, this approach is just not practical."
Machine learning has great potential to counter this challenge, but Acar says the complex way peptides assemble and disassemble has prevented artificial intelligence methods from being effective so far.
"Clearly, computational methods, such as machine learning, are necessary," she said. "Yet, the peptide aggregation is very complex. It is currently not possible to identify the effects of individual amino acids with computational methods."
To counter those challenges, the research team came up with a new approach. They developed a framework that would help bridge materials science and engineering research with computational science to lay the groundwork for artificial intelligence and machine learning advancements.
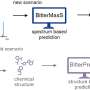
"For this paper, we focused on small peptides with six amino acids, where even still the possible combinations are incredible," Acar said. "We wanted to see what sort of interactions would affect the end product in what way, so we created a framework that kept four of the six amino acids the same all of the time, and we changed the remaining two of them at a time to see how that will affect the interactions and also the product that will come together in the end."
The researchers focused on peptide-aggregation into one-dimensional structures and countered the challenge of the kinetic threshold—where the intermolecular interactions between small peptides are not enough to aggregate in low concentrations—by having the two variable amino acids of the framework hold opposite electron charges.
"In Alzheimer's Disease, all of the amyloids come together in a one-dimensional structure," Acar said. "Therefore, we have tremendous amounts of standardized quantification techniques so we know we can compare them with anything that has been synthesized and published. It makes it comparable with the literature."
"The advantage of this framework is, it is simple enough to make computational simulations, which provide an opportunity to use machine learning," White added.
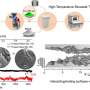
White used molecular dynamics calculations to simulate what happens on the scale of atoms during the initial steps of self-assembly.
"Molecular dynamics enables us to see how these peptides interact, a literal movie of how the atoms line-up to start the self-assembly process," he said.
Through the framework described in their article, the research team lays the groundwork for demonstrating efficacy with six amino acid structures that can be expanded on in later work.
"The data that we will collect in future studies using this framework, although they might not be as we predict now, will give us an important insight on how peptides come together and will likely change the future perspective of peptide engineering," Hamsici said.
Likewise, White says future applications of their research will be able to use deep learning and artificial intelligence to model peptide structures to create materials with needed properties for a desired application.
Acar imagines a future where a materials engineer could input parameters for desired material and a computer simulation could determine the peptide structure that would be needed.
"This framework is a breakthrough in helping experimental scientists and computational scientists to have the same tool to look at the problem from different directions," Acar said. "We want to continue creating different peptides and different properties until we have enough data to build a databank for different materials science to really benefit from machine learning. The technology we described in this paper can pave the way for materials genome atlas for peptide-based materials."
More information: Seren Hamsici et al, Peptide framework for screening the effects of amino acids on assembly, Science Advances (2022). DOI: 10.1126/sciadv.abj0305
Journal information: Science Advances
Provided by University of Oklahoma