Cystic fibrosis drugs can be 'life-changing' for patients: New images reveal how the molecules work
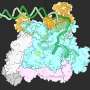
 nestle in a cavity in the protein that malfunctions in cystic fibrosis, stabilizing the protein and correcting its flaws. Credit: K. Fiedorczuk et al, Cell 2022")
A mutated protein causes the genetic disease cystic fibrosis. Now, researchers have solved the structure of the protein and how certain drugs bind to it, unveiling their mode of action.
Just over a decade ago, in one of the great advances in modern medicine, researchers at Aurora Biosciences and then Vertex Pharmaceuticals found what they described at the time as needles in a haystack—small molecule drugs that can treat cystic fibrosis, the lung-damaging genetic disease.
Vertex's first drug, ivacaftor, was approved in 2012, and others followed, dramatically increasing survival age and quality of life for cystic fibrosis patients. The drugs improve lung function, allowing patients to breath better. Scientists knew that the drugs somehow adjusted or corrected the faulty protein responsible for the disease, but how they work has remained unknown.
Now, in new work that reveals the structure of the mutated protein and how so-called corrector drugs interact with it, Howard Hughes Medical Institute Investigator Jue Chen's team at the Rockefeller University has discovered how the molecules operate. They bind to and stabilize the defective protein during its biogenesis, giving it a better chance to successfully reach the cell surface, her team reports January 6, 2022, in the journal Cell.
While many scientists have made strides toward understanding these drugs' mechanism, Chen's new work "is a substantial contribution—it is groundbreaking for the field," says Garry Cutting, a medical geneticist at the Johns Hopkins University School of Medicine who was not involved with the work.
"It is satisfying to see that although these drugs were discovered empirically, they act through basic thermodynamic principles that underlie protein stability," Chen says. "I am also hoping that these data will guide new therapy development to help people who do not respond to current treatments."
Black sheep
Cystic fibrosis is a genetic disease that leads to lung damage, digestive problems, and infections, among other medical problems. It is caused by mutations in a protein called the cystic fibrosis transmembrane conductance regulator (CFTR). Normally, this protein acts as a channel that allows vital ions like chloride to pass through cell membranes. When mutated, CFTR malfunctions or is almost completely absent, leading to an average life expectancy for cystic fibrosis patients of only 30 years or less. Two types of drugs, called potentiators and correctors, have almost doubled that. "The fact that the drugs can tackle the root cause of the disease in 90 percent of patients worldwide is pretty spectacular—life-changing for many patients," Cutting says.
But the scientific puzzle of how a small molecule could correct the defects caused by the mutated protein remained. Chen decided to take on the challenge. She'd already built a successful career illuminating the molecular machinery of so-called ABC transporters—the proteins that pump substances, such as sugars, across cell membranes. But she also had kept her eye on one especially unusual transporter, CFTR. "It's the black sheep in the family because it's the only one that is a channel rather than a pump," she explains. In other words, instead of actively transporting substances from areas of low concentrations to those of higher concentrations, CFTR simply allows molecules to flow down the concentration gradient, like water running downhill. "That made me very interested in this protein," Chen says.
At first, figuring out the structure of the protein—and where the drugs might bind—was virtually impossible. The reason: CFTR is unstable even when not mutated, with parts that flop around. The standard technique, X-ray crystallography, wasn't up to the task. X-ray crystallography relies on proteins that can form a stable crystal structure—something that's not easy with floppy proteins. "You can't get a crystal because parts of the protein move a lot," Chen explains.
That barrier, however, was swept away by a technical breakthrough in 2014, cryogenic electron microscopy. By freezing samples of proteins under study, cryo-EM allowed detailed electron microscope pictures of protein structure and started what has been dubbed the "resolution revolution." Chen first used the technique to study the structures of CFTR in its channel-closed and channel-open forms, published in Cell in 2016 and 2017. Then her team discovered that potentiator cystic fibrosis drugs, which improve the function of mutated CFTR proteins, bind to the protein in a way that keeps the channel open, as reported in Science in 2019.
Unexpected hole
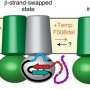
Chen and a postdoctoral associate, Karol Fiedorczuk, then turned to the other type of drug—those that improve the folding of mutated CFTR protein. By taking thousands of electron microscope pictures, the scientists showed that CFTR has an unexpected cavity in its 3D structure. These corrector drugs nestle precisely in that hole, stabilizing the protein and ensuring that it folds into the right shape. "It is surprising until you actually see it, because there are no other examples in the literature of how small molecules can help protein folding," says Chen.
Fiedorczuk and Chen used the 3D structure to propose how molecular glitches lead to cystic fibrosis, and how the drugs come to the rescue. Their idea centers on CFTR's stability—or lack of it.
CFTR is a large complex protein, with five distinct domains. It takes up to two hours for the cell to make and properly fold the protein. That's a lot of time for things to go awry, Chen says. And the cavity her team discovered doesn't help. It occurs even in normal CFTR proteins, making them unstable and prone to degradation. Even under the best of circumstances, 80 percent or more of the typical CFTR protein that cells produce never makes it to the membranes where it's needed.
Cystic fibrosis mutations make the problem even worse. The genetic glitches increase the protein's instability, so virtually none of it ends up where it needs to be, causing the disease. But by binding in the cavity, the drug stabilizes the first part of the protein made by the cell, keeping it from being degraded. That buys the cell enough time to finish making the rest of the protein and fold it into the right shape.
The findings validate an idea, first proposed 30 years ago, that holes in proteins make them unstable, with instability increasing as the holes get bigger, says Chen. "Intellectually, that is very satisfying," she says. More practically, it explains why a single drug can help cystic fibrosis patients with different mutations.
There are still more details to be uncovered, such as where the drugs linger in cells before binding to the protein, says Stephen White, a physiologist and biophysicist at the University of California, Irvine, but he calls Chen's new work "an important starting point."
More information: Karol Fiedorczuk et al, Mechanism of CFTR correction by type I folding correctors, Cell (2022). DOI: 10.1016/j.cell.2021.12.009
Provided by Howard Hughes Medical Institute