Drug discovery: First highly scalable method to monitor protein levels and localizations

Until now, scientists typically studied the changes of proteins and their roles in the cell by using a fluorescent tag to label and follow one protein at a time. This approach limited the number of proteins that could be studied and precluded unbiased discovery approaches. Researchers at CeMM, the Research Center for Molecular Medicine of the Austrian Academy of Sciences, have now developed a highly scalable method which allows for the study of hundreds of proteins in parallel in order to monitor the changes of their levels and localization in the cell. This novel strategy is a notable contribution, not only to drug development for future treatments against diseases such as cancer, but also to our general understanding and knowledge of proteome dynamics. Their findings have now been published in the renowned scientific journal Genome Research.
Proteins are large molecules in the cell, and they are required for the structure, function and regulation of the tissues and organs in the body. They are responsible for nearly every task of cellular life and can be as diverse as the functions they serve. Protein levels and their localization within the cell regulate important aspects of many cellular processes and can become important targets for drug treatment. For example, the abundance of proteins can be increased or decreased by intervening therapeutically, by drugs that affect protein production and degradation in the cell. Proteins can also move between different cellular compartments, and thereby shift their functions. Other proteins might bind to distinct locations in response to external stimuli, such as areas where DNA damage occurs.
Traditionally, scientists use a fluorescent tag to label individual proteins and study their roles in the cell. A green fluorescent protein (GFP) is fused to one of the ends of a certain protein they wanted to study. This protein fusion is then expressed in the cell, and through fluorescence microscopy they can observe the cells expressing the labeled protein. This method permits studying many perturbations like different drug doses in a time resolved manner for a single protein. In contrast, mass-spectrometry was not suitable to study and monitor these cellular perturbations on the proteome, the entire complement of proteins, at a high scalable level on a specific point in time in an unbiased way.
Andreas Reicher and Anna Koren from CeMM Principal Investigator Stefan Kubicek's group have developed a novel strategy, which allows, for the first time, to observe and characterize those changes in a very high number of proteins in parallel. This method can be used to, not only describe and better understand the effects of certain known drugs in the cells, but also to discover new drug treatments that work by affecting and modulating the protein levels or localizations in the cells.
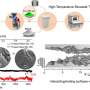
CeMM researchers have designed a method to overcome the bottle-neck in CRISPR-CAS9-based intron tagging: that there is a need to develop methods that shine a light on the whole proteome, or a substantial part thereof and not just one protein at a time. To overcome this problem, CeMM researchers designed a method to generate cell pools containing hundreds of tagged proteins, and in each cell a different protein was labeled with GFP. These cell pools were exposed to a PROTAC chemical degrader of BRD4, a transcriptional regulator that plays a key role during embryogenesis and cancer development.
Researchers then using time-lapse microscopy observed if there were any changes in the levels or subcellular localization of any of the tagged proteins in the cell pool in response to the applied treatment. Importantly, the CRISPR-Cas9 tagging strategy they applied then enabled them to identify which proteins changed localization by using in situ sequencing of the entire cell pool. Thus, they confirmed the known targets of these drug but also revealed unexpected changes. Particularly for perturbations of BRD4 signaling, they were able to report changes in localizations of six proteins that had previously not being recognized by any other high throughput methods. Finally, they also showed that the method reveals expected and novel protein localization changes as response to treatment with the approved cancer drug methotrexate.
CeMM Principal Investigator Stefan Kubicek explains, "Our study describes a technology which, not only, for the first time, applies intron tagging to a gene pool, but is also significantly optimized in all three steps—intron tagging, cellular imaging and in situ sequencing—to enable the process in the most effective way. This method applied to chemical libraries and candidate molecules is particularly powerful in order to develop and deeply characterize drugs including the induction and inhibition of protein-protein interactions and chemical degradation. The described strategy will potentially accelerate drug discovery, and have great impact on the study of global and subcellular proteome dynamics."
More information: Genome Research (2020). DOI: 10.1101/gr.261503.120
Journal information: Genome Research
Provided by Austrian Academy of Sciences