Researchers map protein motion
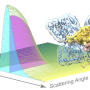
 and the triclinic unit cell containing one protein (bottom). b A highly detailed three-dimensional map of diffuse scattering was obtained. The outer sphere is drawn at 2 Å resolution. c The total scattering is made up of three components: inelastic Compton scattering (lower left), a broad isotropic ring that dominates the diffuse signal (upper left), and variational features in the diffuse scattering (right). Intense halos are visible in the layers containing Bragg peaks (l = 0 plane, upper right). Cloudy scattering is best visualized in the planes mid-way between the Bragg peaks (l = 1∕2 plane, lower right). Credit: Nature Communications (2020). DOI: 10.1038/s41467-020-14933-6")
Cornell structural biologists took a new approach to using a classic method of X-ray analysis to capture something the conventional method had never accounted for: the collective motion of proteins. And they did so by creating software to painstakingly stitch together the scraps of data that are usually disregarded in the process.
Their paper, "Diffuse X-ray Scattering from Correlated Motions in a Protein Crystal," published March 9 in Nature Communications.
As a structural biologist, Nozomi Ando, M.S. '04, Ph.D. '08, assistant professor of chemistry and chemical biology, is interested in charting the motion of proteins, and their internal parts, to better understand protein function. This type of movement is well known but has been difficult to document because the standard technique for imaging proteins is X-ray crystallography, which produces essentially static snapshots.
"Because we're studying really challenging biological systems, the group often has to pioneer new structural methods as well," said postdoctoral researcher Steve Meisburger, Ph.D. '14, the paper's lead author. "One of the questions that we have been interested in since the beginning is how a protein's subtle breathing motions direct biochemical function."
The researchers brought their project to the Cornell High Energy Synchrotron Source (CHESS), where they took advantage of the facility's Pilatus 6M pixel-array detector, which enabled them to make very high-resolution images.
For this work, as in regular crystallography, X-rays were beamed at a sample crystal. The pixel-array detector recorded the intensity of the X-rays that were diffracted by the crystal's proteins, thus encoding the atomic structure. Any disorder—i.e., motion—inside the crystal caused additional photons to bounce out, creating a very weak background signal called diffuse scattering. This information has been traditionally discarded during data processing.
"The photons go everywhere, and the signal appears extremely weak because it's spread out," said Ando, the paper's senior author. "For decades, people couldn't measure it accurately, and they didn't know how to interpret it."
Meisburger created software to process the approximately 50 million unique data points, resulting in a high-quality three-dimensional map. Much to the researchers' surprise, the map revealed that a significant component of this diffuse scattering pattern was actually a result of the protein lattice vibrating. This jiggling movement was so dominant, it seemed to obscure any motion inside the proteins, which was initially a disappointment for the researchers.
But after accounting for these lattice vibrations in simulations, the researchers identified internal protein motions as well. These motions included the opening and closing of the active site of the protein.
"Imagine the crystal being like a row of people trying to walk together while holding hands, but at the same time, each individual might be doing something slightly different," Ando said. "The signal from everybody moving together is dominant, so we couldn't discern the subtle signal that was coming from the individuals. That was something that had never been accounted for."
This new approach to diffuse scattering could help researchers get a clearer picture of protein structure and dynamics and, ultimately, a better understanding of biochemical reactions.
"We really want to push this in a direction where lots of people can use the technique and learn something new about their protein," Meisburger said. "One great thing about it is you get diffuse scattering for free anytime you do a regular crystallography experiment. This technique really adds information to what you would normally get."
More information: Steve P. Meisburger et al. Diffuse X-ray scattering from correlated motions in a protein crystal, Nature Communications (2020). DOI: 10.1038/s41467-020-14933-6
Journal information: Nature Communications
Provided by Cornell University