Supercomputers help supercharge protein assembly

Red blood cells are amazing. They pick up oxygen from our lungs and carry it all over our body to keep us alive. The hemoglobin molecule in red blood cells transports oxygen by changing its shape in an all-or-nothing fashion. Four copies of the same protein in hemoglobin open and close like flower petals, structurally coupled to respond to each other. Using supercomputers, scientists are just starting to design proteins that self-assemble to combine and resemble life-giving molecules like hemoglobin. The scientists say their methods could be applied to useful technologies such as pharmaceutical targeting, artificial energy harvesting, 'smart' sensing and building materials, and more.
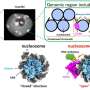
A science team did this work by supercharging proteins, which means that they changed the subunits of proteins, the amino acids, to give the proteins an artificially high positive or negative charge. Using proteins derived from jellyfish, the scientists were able to assemble a complex sixteen protein structure composed of two stacked octamers by supercharging alone, findings that were reported in January of 2019 in the journal Nature Chemistry.
The team then used supercomputer simulations to validate and inform these experimental results. Supercomputer allocations on Stampede2 at the Texas Advanced Computing Center (TACC) and Comet at the San Diego Supercomputer Center (SDSC) were awarded to the researchers through XSEDE, the Extreme Science and Engineering Discovery Environment funded by the National Science Foundation (NSF).
"We found that by taking proteins that don't normally interact with each other, we can make copies that are either highly positively or highly negatively charged," said study co-author Anna Simon, a postdoctoral researcher in the Ellington Lab of UT Austin. "Combining the highly positively and negatively charged copies, we can make the proteins assemble into very specific structured assemblies," Simon said. The scientists call their strategy 'supercharged protein assembly,' where they drive defined protein interactions by combining engineered supercharged variants.
"We exploited a very well-known and basic principle from nature, that opposite charges attract," added study co-author Jens Glaser. Glaser is an assistant research scientist in the Glotzer Group, Department of Chemical Engineering at the University of Michigan. "Anna Simon's group found that when they mix these charged variants of green fluorescent protein, they get highly ordered structures. That was a real surprise," Glaser said.
The stacked octamer structure looks like a braided ring. It's composed of 16 proteins—two intertwined rings of eight that interact in very specific, discreet patches. "The reason why it's so hard to engineer proteins that interact synthetically is that making these interacting patches and having them all line up right such that they'll allow the proteins to assemble into bigger, regular structures is really hard," explained Simon. They got around the problem by adding many positive and negative charges to engineer variants of green fluorescent protein (GFP), a well-studied 'lab mouse' protein derived from the Aequorea victoria jellyfish.
The positively charged protein, which they called cerulean fluorescent protein (Ceru) +32, had additional opportunities to interact with the negatively charged protein GFP -17. "By giving these proteins all these opportunities, these different places where they could potentially interact, they were able to choose the right ones," Simon said. "There were certain patterns and interactions that were there, available, and energetically favored, that we didn't necessarily predict beforehand that would allow them to assemble into these specific shapes."
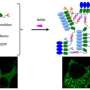
To get the engineered charged fluorescent proteins, Simon and co-authors Arti Pothukuchy, Jimmy Gollihar, and Barrett Morrow encoded their genes, including a chemical tag used for purification on portable pieces of DNA called plasmids in E. coli, then harvested the tagged protein that E. coli grew. The scientists mixed the proteins together. They initially thought the proteins might just interact to form large, irregularly structured clumps. "But then, what we kept on seeing was this weird, funny peak around 12 nanometers, that was a lot smaller than a big clump of protein, but significantly bigger than the single protein," Simon said.
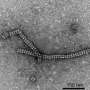
They measured the size of the particles that formed using a Zetasizer instrument at the Texas Materials Institute of UT Austin, and verified that the particles contained both cerulean and GFP proteins Förster Resonance Energy Transfer (FRET), which measures the energy transfer between different colored fluorescent proteins produce fluorescence in response to different energies of light to see if they are close together. Negative stain electron microscopy identifed the specific structure of the particles, conducted by the group of David Taylor, assistant professor of molecular biosciences at UT Austin. It showed that the 12 nm particle consisted of a stacked octamer composed of sixteen proteins. "We found that they were these beautifully shaped flower-like structures," Simon said. Co-author Yi Zhou from Taylor's group of UT Austin increased the resolution even further using cryo-electron microscopy to reveal atomic-level details of the stacked octamer.
 and the Stampede2 supercomputer at the Texas Advanced Computing Center (right). Credit: SDSC, TACC")
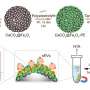
Computational modeling refined the measurements of how the proteins were arranged into a clear picture of the beautiful, flower-like structure, according to Jens Glaser. "We had to come up with a model that was complex enough to describe the physics of the charged green fluorescent proteins and present all the relevant atomistic details, yet was efficient enough to allow us to simulate this on a realistic timescale. With a fully atomistic model, it would have taken us over a year to get a single simulation out of the computer, however fast the computer was," Glaser said.
They simplified the model by reducing the resolution without sacrificing important details of the interactions between proteins. "That's why we used a model where the shape of the protein is exactly represented by a molecular surface, just like the one that's measured from the crystallographic structure of the protein," Glaser added.
"What really helped us turn this around and improve what we were able to get out of our simulations was the cryo-EM data," said Vyas Ramasubramani, a graduate student in chemical engineering at the University of Michigan. "That's what really helped us find the optimal configuration to put into these simulations, which then helped us validate the stability arguments that we were making, and hopefully going forward make predictions about ways that we can destabilize or modify this structure," Ramasubramani said.
The scientists required lots of compute power to do the calculations on the scale that they wanted.
"We used XSEDE to basically take these huge systems, where you have lots of different pieces interacting with each other, and calculate all of this at once so that when you start moving your system forward through some semblance of time, you could get an idea for how it was going to evolve on somewhat real timescales," Ramasubramani said. "If you tried to do the same kind of simulation that we did on a laptop, it would have taken months if not years to really approach understanding whether or not some sort of structure would be stable. For us, not being able to use XSEDE, where you could use essentially 48 cores, 48 compute units all at once to make these calculations highly parallel, we would have been doing this much slower."
The Stampede2 supercomputer at the TACC contains 4,200 Intel Knights Landing and 1,736 Intel Skylake X compute nodes. Each Skylake node has 48 cores, the basic unit of a computer processor. "The Skylake nodes of the Stampede2 supercomputer were instrumental in achieving the performance that was necessary to compute these electrostatic interactions that act between the oppositely-charged proteins in an efficient manner," Glaser said. "The availability of the Stampede2 supercomputer was at just the right point in time for us to perform these simulations."
Initially, the science team tested their simulations on the Comet system at the SDSC. "When we were first figuring out what kind of model to use and whether this simplified model would give us reasonable results, Comet was a great place to try these simulations," Ramasubramani said. "Comet was a great testbed for what we were doing."
Looking at the bigger scientific picture, the scientists hope that this work advances understanding of why so many proteins in nature will oligomerize, or join together to form more complex and interesting structures.
"We showed that there doesn't need to be a very specific, pre-distinguished set of plans and interactions for these structures to form," Simon said. "This is important because it means that maybe, and quite likely we can take other sets of molecules that we want to make oligomerize and generate both positively charged and negatively charged variants, combine them, and have specifically ordered structures for them."
Natural biomaterials like bone, feathers, and shells can be tough yet lightweight. "We think supercharged protein assembly is an easier way to develop the kind of materials that have exciting synthetic properties without having to spend so much time or having to know exactly how they're going to come together beforehand," Simon said. "We think that will accelerate the ability to engineer synthetic materials and for discovery and exploration of these nanostructured protein materials."
The study, "Supercharging Enables Organized Assembly of Synthetic Biomolecules," was published in the journal Nature Chemistry in January of 2019.
More information: Anna J. Simon et al, Supercharging enables organized assembly of synthetic biomolecules, Nature Chemistry (2019). DOI: 10.1038/s41557-018-0196-3
Journal information: Nature Chemistry
Provided by University of Texas at Austin