Modified deep-learning algorithms unveil features of shape-shifting proteins
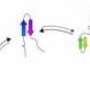
, various transitional forms (2–8), and one misfolded state (9). By studying these protein folding pathways, scientists hope to identify underlying factors that affect human health. Credit: Oak Ridge National Laboratory")
Using artificial neural networks designed to emulate the inner workings of the human brain, deep-learning algorithms deftly peruse and analyze large quantities of data. Applying this technique to science problems can help unearth historically elusive solutions.
One such challenge involves a biophysical phenomenon known as protein folding. Although researchers know that proteins must morph into specific 3-D shapes via this process to function properly, the intricacies of intermediate stages between the initial unfolded state and the final folded state are both critically important to their eventual purpose and notoriously difficult to characterize.
Researchers at the US Department of Energy's (DOE's) Oak Ridge National Laboratory (ORNL) employed a suite of deep-learning techniques to identify and observe these temporary yet notable structures. They published their findings in BMC Bioinformatics.
The team adapted an existing deep-learning algorithm known as a convolutional variational autoencoder (CVAE), which automatically extracted relevant information about protein folding configurations from molecular dynamics (MD) simulations. The researchers ran these simulations on Summitdev, a small-scale precursor to Summit, currently the world's most powerful supercomputer, which is located at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science User Facility at ORNL.
By studying the folding pathways of three different proteins—namely Fs-peptide, villin head piece, and BBA—the researchers computationally compared multiple protein folding mechanisms. They relied on datasets obtained from other research groups that have run extensive simulations to examine these pathways. In each case, the CVAE revealed many intermediate stages that serve as "guideposts" to help the team navigate the folding process from start to finish while observing latent facets of protein behavior.
"We took the protein folding trajectories compiled from running MD simulations and fed them into the deep-learning network, which automatically uncovered the relevant guideposts for various proteins," said Arvind Ramanathan, a former ORNL researcher who led this effort.
"These relevant guideposts are picked in a completely unsupervised manner from the high dimensional folding trajectories in such a way that only biophysically relevant features important to that particular system are chosen," added ORNL computational scientist Debsindhu Bhowmik, who implemented the CVAE algorithm customized for the protein systems.
Ramanathan compared this ability to pinpoint transitional protein states to a driver choosing logical pitstops en route from one region to another.
"If you are driving from Knoxville in East Tennessee all the way to Memphis in West Tennessee, then the natural stopping point is Nashville," Ramanathan said. "Just as there are many different routes you can take to reach a road trip destination, there are many different paths proteins take to fold into their final shapes."
However, even the most minute change to these folding pathways can cause proteins to "misfold" into dysfunctional shapes. Misfolding is often attributed as a leading factor in the development of diseases including Alzheimer's, cardiovascular disorders, and diabetes.
"The overall shape of a protein determines its function, so some small perturbation in that shape can produce a misfolded protein and lead to serious medical conditions," Ramanathan said.
With this capacity to differentiate between correctly folded and misfolded proteins, the researchers could gain additional insights into why proteins misfold, how other factors contribute to the development of deadly diseases, and which treatment regimens are most likely to prevent or cure them. For example, identifying a problematic site in a particular protein might indicate the need for planting a binding agent or drug to change that protein's behavior.
Reaching this goal will require increasingly precise techniques, which the team hopes to develop by modeling multiple machine-learning algorithms on NVIDIA DGX-2 boxes, computing systems that enable novel artificial intelligence applications. The DGX-2s were recently installed at ORNL's Compute and Data Environment for Science (CADES), which provides ORNL staff with the infrastructure and expertise needed to complete data-intensive projects.
The researchers focused on optimizing reinforcement-learning algorithms, which perform tasks without preliminary training, then steadily learn from experience to maximize rewards and minimize negative outcomes. One prominent example, Google's AlphaGo computer program, defeated a world champion in the board game Go. Similar reinforcement-learning algorithms are also embedded in arcade and console video games, and the team plans to customize this method for scientific purposes, including gathering and interpreting protein folding data.
"One way to steer MD simulations is to use these powerful reinforcement-learning techniques, but adapting them for these types of simulations requires quite a bit of work and computing power," Ramanathan said.
To improve the algorithms, the team had to optimize hyperparameters, which are parameters set before algorithms start making decisions. Running multiple algorithms on the DGX-2s at once allowed the team to quickly compile data they used to develop HyperSpace, a specialized software package that simplifies and streamlines the process of hyperparameter optimization.
The researchers presented this work at the 2018 High Performance Machine Learning Workshop, an annual event where machine learning, artificial intelligence, and high-performance computing experts gather to discuss experiences and share expertise.
"We found that, for a variety of machine-learning algorithms such as deep-learning algorithms, convolutional neural networks, and reinforcement-learning algorithms, HyperSpace is quite successful and outperforms comparable models," Ramanathan said.
Now, the scientists are building a scalable workflow to benefit future research involving protein folding and other biological phenomena, some of which they plan to study on Summit.
"Although we have focused mostly on protein folding so far, we are actively probing other questions such as how two separate proteins interact with each other," Ramanathan said.
More information: Debsindhu Bhowmik et al. Deep clustering of protein folding simulations, BMC Bioinformatics (2018). DOI: 10.1186/s12859-018-2507-5
Provided by Oak Ridge National Laboratory