Scientists decode dynamics of the largest protein-degrading machine in atomic detail

Protein nanomachines made of multiple protein molecules are highly dynamic during their actions on their functional targets, sometime called substrates. Dynamics of these large protein nanomachines of more than megadalton molecular weight are refractory to structural analysis by existing technology like X-ray crystallography and nuclear magnetic resonance spectroscopy. Cryo-electron microscopy (cryo-EM), an emerging technology for high-resolution structure determination, has potential to visualize dynamics of large protein nanomachines, but the existing cryo-EM reconstructions of highly dynamic structures have been limited to moderate to low resolution.
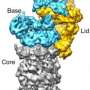
Scientists have long dreamed of decoding dynamics of large molecular machines of megadalton sizes in atomic detail, the ultimate determinant of their biological functions. Now, a team of biophysicists from Peking University, Dana-Farber Cancer Institute and Harvard Medical School have used cryo-EM to visualize atomic-level dynamics of the 2.5-megadalton proteasome, the largest known protein-degrading machine in eukaryotic cells, during its chemo-mechanical action on a protein substrate. They reconstructed a nearly complete dynamic procedure of substrate processing in the human proteasome at unprecedented resolution that allowed the determination of atomic details in 3-D, akin to filming a 3-D movie atom by atom.
"This work paves the way to study thermodynamics of megadalton nanomachines at atomic precision far away from equilibrium," said Youdong Mao, a biophysicist and corresponding author on a new breakthrough paper published in the first issue of the journal Nature in 2019. "This study opens up numerous possibilities for structure-based drug discovery targeting human proteasome for treatment of multiple myeloma and neurodegenerative diseases."
The ubiquitin-proteasome system (UPS) is the most important protein degradation pathway in cells. It maintains the balance of protein materials in living cells, and plays a crucial role in rapid degradation of regulatory proteins, misfolded proteins or damaged proteins. UPS is involved in arguably all cellular processes, such as the cell cycle, gene expression regulation and so on. Abnormal protein metabolism caused by UPS disorder is directly related to many human diseases including cancer. In 2004, Aaron Ciechanover, Irwin Rose and Avram Hershko were awarded the Nobel Prize in chemistry for their discovery of this degradation pathway. At the heart of the UPS is the proteasome responsible for breakdown of ubiquitin-tagged substrates. It is one of the most fundamental and complicated gigantic holoenzyme machines in cells. Human proteasome holoenzyme contains at least 33 different subunit types with a total molecular weight of about 2.5-megadalton. It is also known as the direct target of several small-molecule drugs approved by FDA of United States to treat multiple myeloma.
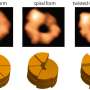
Using cryo-EM in combination with machine learning technology, the team determined dynamic structures of the substrate-engaged human proteasome in seven intermediate conformational states at 2.8-3.6 Å resolution, captured during breakdown of a polyubiquitylated protein. At this resolution, the team was able to identify single magnesium ions bound to both ATP and ADP in the cryo-EM density maps. These 3-D structures illuminate a remarkable spatiotemporal continuum of dynamic substrate-proteasome interactions.
Intriguingly, the team found that the initiation of substrate translocation is extensively coordinated with other dynamic regulatory events preparing the proteasome for processive substrate degradation. Through further systematic analysis, the team discovered how the chemical energy of ATP hydrolysis is converted into the mechanical work of substrate unfolding through a highly concerted process of multi-protein conformational changes.
Their finding provides novel insights into the complete cycle of substrate processing and suggests distinct modes followed by ATP hydrolysis in the proteasome holoenzyme. It is believed to be the first time that a complete cycle of sequential ATP hydrolysis in an AAA-ATPase heterohexameric motor was visualized at the atomic level. This resolves a longstanding scientific debate about ATPase hexamers between two hypothesized models, one suggesting sequential ATP hydrolysis and the other assuming random hydrolytic events in the hexameric ring. Notably, the team observed three principal modes of highly coordinated ATP hydrolysis, featuring hydrolytic events in two oppositely positioned ATPases, in two adjacent ATPases, and in one ATPase at a time. These hydrolytic modes elegantly regulate deubiquitylation, translocation initiation, and processive unfolding of substrates, respectively.
The team noted certain limitations in this study, including that the multiplicity of nucleotide processing events in distinct ATPases during transitions between consecutive states of the proteasome may have resulted in the absence of fast steps and sparsely populated intermediate states in their cryo-EM reconstructions. The team envisions the prospect of further explorations in this regard by identifying these missing intermediaries to clarify how ATP hydrolytic events and nucleotide exchange are coordinated with each other, and allosterically linked to substrate translocation. "Further development in data analysis technology is required to extract even more dynamic information from the same dataset," Mao said. "There is a long way to go for data-driven machine learning technology to fully unleash the potential power of cryo-EM in solving complex dynamics of megadalton molecular machines."
More information: Yuanchen Dong et al, Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome, Nature (2018). DOI: 10.1038/s41586-018-0736-4
Journal information: Nature
Provided by Peking University-College of Engineering