How genetic background shapes individual differences within a species

Study reveals how genetic background influences trait inheritance laying the grounds for predicting personal risk of disease.
Almost every family has a hard-drinking, bacon-loving, exercise-averse relative who despite all odds lived happily to a ripe old age while another perished before their time despite being mindful about their health. Looking at their genomes, it would be impossible to tell who's the lucky one.
This is because of genetic background—countless and mostly subtle differences in the genomes of any two people, which affect gene function in ways scientists don't yet understand.
A new study led by investigators Brenda Andrews and Charles Boone, professors at the University of Toronto's Donnelly Centre for Cellular and Biomolecular Research begins to unpick how genetic background shapes the differences between members of the same species. Andrews is also Director of the Centre and both are professors in the U of T's Department of Molecular Genetics and are Senior Fellows of the Genetic Networks program at the Canadian Institute for Advancement of Research. Boone is also a co-Director of the same CIFAR program. The team also included Gerry Fink, professor of genetics at MIT and member of the Whitehead Institute.
Their findings are published in the journal Proceedings of the National Academy of Science.
"Genetic background confounds our ability to interpret the information stored in an individual genome," says Andrews. It also makes it hard for physicians to predict disease severity in relatively straightforward cases where a disease-causing gene is well known. Two people carrying a same mutation that causes cystic fibrosis, an inherited lung disorder, can develop a mild and a severe form of disease due to the differences between their genetic backgrounds.
With 3 million differences in the DNA code between any two people, the study of genetic background effects in humans is still a daunting prospect. But scientists are beginning to make headway by looking at simpler organisms such as yeast.
"Genetic background has the power to make the original phenotype (a physical outcome of gene function) less or more severe," says Jing Hou, a postdoctoral fellow in the lab who spearheaded the study. This is true for human diseases and it is also true in yeast which is a very good model to study this." This is because the yeast genome is smaller than human and therefore easier to study.
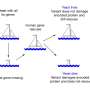
To begin to unpick the genetic background effects, Hou compared how gene mutations manifest themselves in two closely related yeast strains, S288c and sigma1278b, SC and Σ from here, respectively. The two strains are 0.2 per cent different at the DNA level, which is about the same amount of genetic diversity between any two people. In an earlier work, the Boone and Andrews labs, in collaboration with Fink's group, established that mutations in 57 genes, about one percent of all yeast genes, have different outcomes between SC and Sigma, causing cell death in either one or the other strain, but not both. These genes are called "conditional lethals" and whether or not a cell needs them depends on other, so-called modifier genes. But which ones?
By mating the two strains, Hou was able to identify these modifier genes thanks to their ability to mask the damaging mutations and rescue survival of the hybrid progeny.
Hou found that while most conditional lethal genes have multiple modifiers, whose effects are more complex and harder to establish, some have only one modifier and are easier to study. This is the case with CYS3 and CYS4 genes, which help make cysteine, an essential amino acid. Both CYS3 and CYS4 are conditionally lethal in Sigma, but not in the SC strain, which means that Sigma cells die when either gene is missing. Hou discovered that this is thanks to a single modifier gene called OPT1, which works downstream from the CYS genes and can compensate for their loss in the SC strain. Sigma cells happen to carry a mutation in the OPT1 gene and this makes them fully reliant on the CYS genes to produce cysteine.
In another experiment, Hou looked across 20 different yeast strains, from about 1000 naturally found isolates whose genomes have been sequenced. She found a different modifier of the CYS genes in another strain used in the making of Japanese rice wine sake.
With all this information, Hou was able to scan the genomes of all 1000 yeast isolates and accurately guessed which other strains will act like Sigma or the Sake yeast and be completely reliant on the CYS genes to survive. This is similar to being able to single out, from 1000 patients with the same genetic disorder, those individuals who have a higher chance of developing a more severe form of disease.
Being able to predict a biological outcome from genome sequence alone is one of the goals of precision medicine and this early work in yeast raises hopes that similar studies will be possible for human cells.
"Just based on sequence and the knowledge of this pathway we could predict gene essentiality across the whole species," says Hou. "I think we will be able to predict human risk of disease if we have good enough knowledge of how genes work together in pathways." The studies of human gene interactions are only just beginning, however.
For Hou, the yeast work continues and on a much bigger scale. With Guihong Tan, a research associate in the lab, she is working to identify all the genes across 200 isolates whose effects are modified by the genetic background. Tan thinks this number will be 800 genes, but Hou is more conservative in her estimate. "I think we'll find about 200," she says. It's an uncharted territory and the bets are on, with a bottle of champage as prize that Hou hopes to pop once they collect all the data.
More information: Jing Hou el al., "Complex modifier landscape underlying genetic background effects," PNAS (2019). www.pnas.org/cgi/doi/10.1073/pnas.1820915116
Journal information: Proceedings of the National Academy of Sciences
Provided by University of Toronto