Study shows machine learning can improve catalytic design
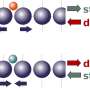
 depicts the charge transfer (blue/green) between metal atoms and an underlying support (orange). This is but one description of a catalyst's physical behavior, and researchers created a massive database by calculating 330,000 such descriptions for each of many catalysts. Machine learning was used (upper panel) to search the database for hidden patterns that designers can use to make cheaper, more efficient catalysts. Credit: Tom Senftle/Rice University")
Chemical engineers at Rice University and Pennsylvania State University have shown that combining machine learning and quantum chemistry can save time and expense in designing new catalysts.
"Large amounts of data are generated in computational catalysis, and the field is starting to realize that data science tools can be extremely valuable for sifting through high-volume data to look for fundamental correlations that we might otherwise miss," said Rice's Thomas Senftle, co-author of a new study published online this week in Nature Catalysis. "That's what this paper was really about. We combined well-established tools for data generation and analysis in a way that allowed us to look for correlations we wouldn't otherwise have noticed."
A catalyst is a substance that accelerates chemical reactions without being consumed by them. The catalytic converters in automobiles, for example, contain metals like platinum and palladium that aid in reactions that break down air pollutants. Catalysts are a mainstay of the chemical and pharmaceutical industries, and the global market for catalysts is estimated at $20 billion per year.
The metals used in catalytic converters are typically part of a wire mesh. As hot exhaust passes through the mesh, the metal atoms on the surface catalyze reactions that break apart some noxious molecules into harmless byproducts.
"That's a gas phase reaction," Senftle said of the catalytic converter example. "There's a certain concentration of gas-phase species that come out of the engine. We want a catalyst that converts pollutants into harmless products, but different cars have different engines that put out different compositions of those products, so a catalyst that works well in one situation may not work as well in another."
The practice of flowing reactants past a catalyst is also common in industry. In many cases, a catalytic metal is attached to a solid surface and reactants are flowed over the surface, either as a liquid or a gas. For industrial processes that make tons of products per years, improving the efficiency of the metal catalyst by even a few percent can translate into millions of dollars for companies.
"If you have a clear picture of the properties of the metal catalyst and the substrate material the metal attaches to, that allows you to basically narrow down your search at the beginning," Senftle said. "You can narrow your design space by using the computer to explore which materials are likely to do well under certain conditions."
Senftle, assistant professor in chemical and biomolecular engineering at Rice, began the newly published research while still a graduate student at Penn State in 2015, along with lead authors Nolan O'Connor and A.S.M. Jonayat and co-author Michael Janik. They started by using density functional theory to calculate the binding strengths of single atoms of many different kinds of metals with a range of metal oxide substrates.
"The binding energy between the metal and substrate is of particular interest because the stronger the bond, the less likely the metal atom is to dislodge," Janik said. "If we can control that binding energy, we can tailor the size distribution of these metal particles, and that, in turn, is going to impact the overall reaction that they can catalyze."
O'Connor said, "We were curious about the properties of individual metal atoms and oxide surfaces that made for strong interacting pairs, which is a property we can use to design robust catalysts."
Along with the list of binding energies, the team had a catalog of about 330,000 additional properties for each of the metal-substrate combinations, including factors like oxide formation energy, coordination number, alloy formation energy and ionization energy.
"The machine learning algorithm looks for the combinations of those descriptors that correlate with the observed data on binding energies," Jonayat said. "It basically allows us to ask, 'Of all of these descriptors, how can we find the ones that correlate with the observed behavior in which we're interested?"
He said identifying such correlations can streamline catalyst design by making it possible to predict how materials will behave prior to laboratory testing that can be both expensive and time-consuming. Machine learning also can identify interesting effects that are worthy of additional study.
For example, Senftle said one correlation that kept appearing in the study was the importance of the direct interaction between the catalytic metals and the metal atoms in the support. He said this was unexpected because the metals typically each have a strong affinity to bind with oxygen as opposed to binding with each other.
"Originally, the idea was that it was the oxygen that was important," Senftle said. "We were interested in determining how well these two different metals shared the oxygen. But this direct interaction between the metals themselves kept popping up in our calculations, and it played a much larger role in dictating the overall behavior of the system than we had anticipated."
Senftle said he'd like to build on the complexity of the simulations in future research.
"Here we were looking at the interactions between the metals and the supports in a pristine environment with no water molecules or impurities of any kind," he said. "In reality, catalysts are used in very complicated reaction environments, and we'd like to examine how these trends change in those settings. For example, if this were an aqueous environment, water or dissociated water would likely adsorb on the surface. Those are going to impact the interaction, because now you have another player that is sharing the electron density and sharing the surface oxygens."
More information: Nolan J. O'Connor et al, Interaction trends between single metal atoms and oxide supports identified with density functional theory and statistical learning, Nature Catalysis (2018). DOI: 10.1038/s41929-018-0094-5
Journal information: Nature Catalysis
Provided by Rice University