Discovery lights path for Alzheimer's research

A probe invented at Rice University that lights up when it binds to a misfolded amyloid beta peptide—the kind suspected of causing Alzheimer's disease—has identified a specific binding site on the protein that could facilitate better drugs to treat the disease.
Even better, the lab has discovered that when the metallic probe is illuminated, it catalyzes oxidation of the protein in a way they believe might keep it from aggregating in the brains of patients.
The study done on long amyloid fibrils backs up computer simulations by colleagues at the University of Miami that predicted the photoluminescent metal complex would attach itself to the amyloid peptide near a hydrophobic (water-avoiding) cleft that appears on the surface of the fibril aggregate. That cleft presents a new target for drugs.
Finding the site was relatively simple once the lab of Rice chemist Angel Martí used its rhenium-based complexes to target fibrils. The light-switching complex glows when hit with ultraviolet light, but when it binds to the fibril it becomes more than 100 times brighter and causes oxidation of the amyloid peptide.
"It's like walking on the beach," Marti said. "You can see that someone was there before you by looking at footprints in the sand. While we cannot see the rhenium complex, we can find the oxidation (footprint) it produces on the amyloid peptide.
"That oxidation only happens right next to the place where it binds," he said. "The real importance of this research is that allows us to see with a high degree of certainty where molecules can interact with amyloid beta fibrils."
The study appears in the journal Chem.
"We believe this hydrophobic cleft is a general binding site (on amyloid beta) for molecules," Martí said. "This is important because amyloid beta aggregation has been associated with the onset of Alzheimer's disease. We know that fibrillar insoluble amyloid beta is toxic to cell cultures. Soluble amyloid oligomers that are made of several misfolded units of amyloid beta are also toxic to cells, probably even more than fibrillar.
"There's an interest in finding medications that will quench the deleterious effects of amyloid beta aggregates," he said. "But to create drugs for these, we first need to know how drugs or molecules in general can bind and interact with these fibrils, and this was not well-known. Now we have a better idea of what the molecule needs to interact with these fibrils."

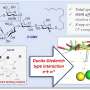
When amyloid peptides fold properly, they hide their hydrophobic residues while exposing their hydrophilic (water-attracting) residues to water. That makes the proteins soluble, Martí said. But when amyloid beta misfolds, it leaves two hydrophobic residues, known as Valine 18 and Phenylalanine 20, exposed to create the hydrophobic cleft.
"It's perfect, because then molecules with hydrophobic domains are driven to bind there," Martí said. "They are compatible with this hydrophobic cleft and associate with the fibril, forming a strong interaction."
If the resulting oxidation keeps the fibrils from aggregating farther into the sticky substance found in the brains of Alzheimer's patients, it may be the start of a useful strategy to stop aggregation before symptoms of the disease appear.
"It's a very attractive system because it uses light, which is a cheap resource," Martí said. "If we can modify complexes so they absorb red light, which is transparent to tissue, we might be able to perform these photochemical modifications in living animals, and maybe someday in humans."

He said light activation allows the researchers to have "exquisite control" of oxidation.
"We imagine it might be possible someday to prevent symptoms of Alzheimer's by targeting amyloid beta in the same way we treat cholesterol in people now to prevent cardiovascular disease," Martí said. "That would be wonderful."
More information: Chem (2017). DOI: 10.1016/j.chempr.2017.09.011 , www.cell.com/chem/fulltext/S2451-9294(17)30401-1
Journal information: Chem
Provided by Rice University