Supercomputers model key details of greenhouse gases and molecules relevant to automobile combustion
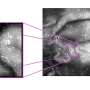
 molecule oscillates faster than a normal 12CH2D2 (left) molecule. Using the new computational method to investigate such fine differences can reveal details relevant to combustion chemistry. Credit: US Department of Energy")
Supercomputers can calculate how a molecule's atoms move, giving insights for astrophysics, fuel use, and more. To determine a molecule's rotational and vibrational energies, chemists collect data about the positions of its atoms in space. The data is a table of numbers. Without a more clever approach to calculating the positions, the largest supercomputers are overwhelmed by six-atom molecules, for which the table would contain hundreds of billions of rows and columns. Thanks to chemists at Brookhaven National Laboratory, supercomputers can now calculate vibrational energies for larger, floppy molecules without relying on approximations.
The new method substantially improves the reliability of comparisons between computed and experimental spectra. These comparisons provide means to ensure the accuracy of the computational method. Calculating how molecules twist and oscillate is important in understanding everything from how the universe works to interpreting new results on combustion engines.
For semi-rigid molecules at low energies, the well-established normal mode approximation gives a basic understanding of molecular vibrations and their associated infrared spectra. For floppy molecules, or molecules with enough vibrational energy to distort far from the low-energy structure, more powerful theoretical tools are required. The problem can be posed without approximation as a large matrix diagonalization, but with an apparently intractable scaling problem. Each additional atom in the molecule requires diagonalization of a matrix with up to 1000 times as many rows and columns as was required for the smaller molecule. Without a more clever approach, this scaling would overwhelm the largest current supercomputers for six-atom molecules, for which the relevant square matrix would have hundreds of billions of rows and columns.
At Brookhaven National Laboratory, computational chemists have developed efficient, rigorous methods to solve the equivalent problem by recasting it as a nested set of lower dimensional problems. The general algorithm has been applied to compute highly excited vibrational frequencies for molecules with up to six atoms. The most recent multi-layer Lanczos algorithm can provide accurate spectroscopic absorption strengths, in addition to the vibrational transition frequencies, at only modest additional computational cost. The method requires an accurate dipole moment surface in addition to the ab initio potential energy surface, but provides an additional key observable for comparison with experiment, to highlight the dominant features in a complex absorption spectrum. An application to 91 experimental infrared vibrational bands of CH2D2 showed excellent frequency agreement (0.67 cm-1 root-mean-square error) and reassigned 20 bands. The team discovered an anomalous carbon isotope shift in the CH2 twisting vibrational mode.
More information: H.G. Yu, "Multi-layer Lanczos iteration approach to calculations of vibrational energies and dipole transition intensities for polyatomic molecules." Journal of Chemical Physics 142, 044106 (2015). DOI: 10.1063/1.490692
Journal information: Journal of Chemical Physics
Provided by US Department of Energy