January 27, 2016 report
Synthetic strategy for enantioselective aldol reactions with fluoroacetate

(Phys.org)—Fluorine, the most electronegative atom on the periodic table, has some interesting applications in organic and medicinal chemistry. Replacing hydrogen with fluorine can often boost biological activity and carbon-fluorine bonds are strong enough to withstand metabolic conditions. To that end, many scientists are interested in making fluorinated derivatives of pharmaceuticals. Unfortunately, it is difficult to functionalize certain organic compounds with fluorine.
Jakub Saadi and Helma Wennemers from the Laboratory of Organic Chemistry at ETH Zurich have developed a general biomimetic method that uses fluoromalonic acid half thioesters as fluoroacetate surrogates, which allows for an enantioselective aldol reaction that selectively places a fluoroacetate on an organic compound. Their work appears in Nature Chemistry.
Acetate is a common building block for many natural produces and pharmaceuticals, including statins and polyketides. One way to incorporate fluorine into a common pharmaceutical would be to use fluoroacetate as a building block since its reactivity would be very similar to acetate. The problem is that fluoroacetate aldol reactions lead to a racemic mixture of products. In pharmaceuticals the "handedness" of a molecule is important for its biocompatibility. Racemic mixtures have both left- and right-handed molecules which are typically not able to be separated. The ideal case would be an enantioselective fluoroacetate reaction.
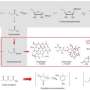
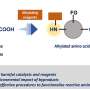
Saadi and Wennemers devised a strategy using Meldrum's acid as a precursor to a reaction that produces fluoromalonic acid half thioesters (F-MAHT). Meldrum's acid is a rigid ring structure. Fluorine can displace the hydrogen between the two carbonyl groups on Meldrum's acid in a three-step process. This compound then undergoes a nucleophilic ring-opening reaction in which it is reacted with a silylated thiophenol to produce the silyl ester intermediate that is hydrolyzed to the F-MAHT.
F-MAHT is used rather than fluoroacetate in an aldol reaction because it allows for mild and selective reaction conditions as well as evolving CO2, thus driving it toward the products. Furthermore, using a quinidine-urea catalyst would allow control over which face of the F-MAHT reacts with an aldehyde. Using this strategy, once Saadi and Wennemers optimized their reaction conditions, they were able to obtain several fluoroaldol products of reactions between F-MAHT derivatives and various aromatic and aliphatic aldehydes with enantioselective control.
They then tested whether their procedure would work for a real life target by making a short total synthesis of a fluorinated analog of atorvastatin (Lipitor) in which they ended up with product (30% overall yield). The key step involved their F-MAHT aldol reaction and reduction to the fluoroaldehyde intermediate. By this they also demonstrated that F-MAHTs can also serve as fluoroacetaldehyde surrogates. Fluoroacetaldehyde is highly reactive and yields complex mixtures of many products. Saadi and Wennemers's method overcomes this problem.
Just as fluorinated versions of several other compounds, such as hydrocortisone, showed enhanced biological activity, the hope is that this mechanism will open the door to many other fluorinated pharmaceuticals with improved properties compared to their non-fluorinated analogs.
More information: Jakub Saadi et al. Enantioselective aldol reactions with masked fluoroacetates, Nature Chemistry (2016). DOI: 10.1038/nchem.2437
Abstract
Despite the growing importance of organofluorines as pharmaceuticals and agrochemicals, the stereoselective introduction of fluorine into many prominent classes of natural products and chemotherapeutic agents is difficult. One long-standing unsolved challenge is the enantioselective aldol reaction of fluoroacetate to enable access to fluorinated analogues of medicinally relevant acetate-derived compounds, such as polyketides and statins. Herein we present fluoromalonic acid halfthioesters as biomimetic surrogates of fluoroacetate and demonstrate their use in highly stereoselective aldol reactions that proceed under mild organocatalytic conditions. We also show that the methodology can be extended to formal aldol reactions with fluoroacetaldehyde and consecutive aldol reactions. The synthetic utility of the fluorinated aldol products is illustrated by the synthesis of a fluorinated derivative of the top-selling drug atorvastatin. The results show the prospects of the method for the enantioselective introduction of fluoroacetate to access a wide variety of highly functionalized fluorinated compounds.
Journal information: Nature Chemistry
© 2016 Phys.org