Researchers develop versatile new way to build molecules
Chemists at The Scripps Research Institute (TSRI) have devised a new and widely applicable technique for building potential drug molecules and other organic compounds.
The new method, reported in the January 15, 2016 issue of the journal Science, enables researchers to add clusters of atoms called carbon fragment or functional groups to certain organic molecules more efficiently, robustly and selectively than current methods typically allow. It thus opens up new possibilities for chemists to assemble novel compounds that can be tested for useful properties in the development of drugs and other products.
"We demonstrated this technique with two broad classes of compounds, aldehydes and ketones—the 'bread and butter' of modern chemical synthesis," said senior investigator Jin-Quan Yu, the Frank and Bertha Hupp Professor of Chemistry at TSRI.
Expanding the Toolkit
Yu's laboratory specializes in the development of techniques to make molecule-building easier, particularly for chemists trying to devise potential new drugs. Yu and his team have published more than a half-dozen of these innovations in Science or Nature in the past two years alone.
Their newest tool improves a basic molecule-building operation called C-H functionalization. When chemists set out to build a candidate drug molecule, they often start with a simple organic compound whose central structure contains more inert carbon hydrogen bonds than reactive carbon heteroatom bonds. Turning such a starter molecule into a useful drug typically means replacing at least one of the hydrogen atoms with a more complex cluster of atoms called a functional group.
This C-H functionalization process can be tricky for a variety of reasons, and chemists often have to employ special methods to make it work. Many of these methods involve helper molecules known as "directing groups." Chemists first attach a directing group to the initial molecule they want to modify; the directing group then guides a bond-breaking catalyst, often a metal such as palladium, to the carbon-hydrogen bond that needs to be broken to make way for the new functional group.
"This has proven to be a very reliable and broadly useful strategy," said Yu, "but it requires at least two additional steps—the installation of the directing group and later its removal—and sometimes the directing group is incompatible with functional groups already present on the starting molecule."
Ideally, chemists would like to find directing groups that are broadly tolerant of existing functional groups and that also don't have to be attached and detached in separate steps. Essentially that is what Yu and his team have achieved here.
Cutting Out Two Steps
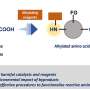
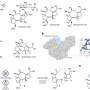
The team—including co-first authors Fang-Lin Zhang, a visiting scholar from Wuhan University of Technology; Kai Hong, a postdoctoral research associate in the Yu Laboratory; and Tuan-Jie Li, a visiting scholar from Jiangsu Normal University—found that amino acid molecules (the building blocks of the proteins that help make up all known life forms) can work well as "transient directing groups" for ketone or aldehyde compounds. The amino acids attach themselves automatically to these starter compounds and remove themselves automatically after the new functional group is attached.
In effect, this means that they work "catalytically," functionalizing one starter molecule after another and continually being re-used, rather than being consumed in their first reaction. This further streamlines the process and reduces the overall quantity of reagents that are needed.
"In principle, all amino acids can be used as catalytic directing groups for such reactions," said Yu. "The availability of diverse amino acids makes it possible to find different reagents to suit different substrates or transformations."
A further advantage of the new technique is that it can, with the proper choice of chiral amino acid directing group, preferentially generate "chiral" molecules that are functionalized just on one side. C-H functionalization reactions typically generate a roughly even mix of molecules functionalized on one side plus mirror-image molecules functionalized on the other side—yet the desirable biological activity of a drug often comes exclusively from its "right-handed" or "left-handed" chiral form.
"The fact that we can do this using a simple amino acid as the directing group and ligand is phenomenal, considering the usual difficulty of such reactions," said Hong.
"Essentially with this new method we're improving functionalizations by cutting out two steps in the functionalization process—by using a directing group that is catalytic, and by employing, if needed, a chiral directing group to generate chirally pure compounds," said Yu.
Yu and his team are now working to extend the applicability of the new method to other broad classes of medicinal chemistry compounds such as amines and alcohols.
More information: "Functionalization of C(sp3)–H bonds using a transient directing group" DOI: 10.1126/science.aad7893
Journal information: Science
Provided by The Scripps Research Institute