Changes to DNA on-off switches affect cells' ability to repair breaks, respond to chemotherapy

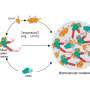
Double-strand breaks in DNA happen every time a cell divides and replicates. Depending on the type of cell, that can be pretty often. Many proteins are involved in everyday DNA repair, but if they are mutated, the repair system breaks down and cancer can occur. Cells have two complicated ways to repair these breaks, which can affect the stability of the entire genome.
Roger A. Greenberg, M.D., Ph.D., associate investigator, Abramson Family Cancer Research Institute and associate professor of Cancer Biology at the Perelman School of Medicine, University of Pennsylvania, together with postdoctoral researcher Jiangbo Tang Ph.D. and colleagues, found a key determinant in the balance between two proteins, BRCA1 and 53BP1, in the DNA repair machinery. Breast and ovarian cancer are associated with a breakdown in the repair systems involving these proteins. Their findings appear in the latest online issue of Nature Structural & Molecular Biology.
The two proteins, BRCA1 and 53BP1, control which of two cell-repair mechanisms will be used: homologous recombination or non-homologous end-joining, technically speaking. This competition has proven to be a key factor in determining whether a cell becomes cancer prone as well as how a cancer cell will respond to chemotherapy.
The key step of the balance is acetylation, the chemical process of adding a compound called an acetyl group to other cellular molecules.
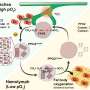
The researchers asked what cell signals determine whether BRCA or 53BP1 predominates at a DNA break site.
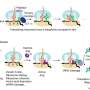
DNA in the nucleus is tightly packed around proteins called histones. Acetylation at a specific spot on histone H4 determines the answer. If H4 is acetylated at a specific location, then 53BP1 binding near the broken DNA region is strongly reduced. This leaves BRCA1 free to do the work, kicking in the homologous recombination tool to repair the break.
On the other hand, if acetylation is reduced, 53BP1 outcompetes BRCA1 at a break and the non-homologous end-joining tool repairs the break.
This mechanism can help explain resistance to a promising chemotherapy called PARP inhibition seen in patients and mouse models with BRCA1 mutations. Work from several other research teams surprisingly has shown that if neither BRCA nor 53BP1 are available, then the homologous recombination system goes into action even in the absence of BRCA1 and BRCA1 mutant cancer cells become resistant to PARP inhibitors.
Because of this, Greenberg says, there are some possible applications for making PARP chemotherapy more sensitive: "If you could inhibit specific acetylation events, then a patient's response to PARP inhibitors might be enhanced by hyperactivating 53BP1 binding to breaks in the context of BRCA1 deficient cancers. What's more, measuring the levels of acetylation at H4 might predict how responsive an individual is to PARP inhibitors."
"The story didn't fall into place the way we thought it would," says Greenberg. "We didn't realize that it was a combination of two epigenetic marks that drives the repair system. However, we were able to show that 53BP1 doesn't bind well to regions of histone H4 that are acetylated at a specific location on H4. Collaboration with Georges Mer, a structural biologist at the Mayo Clinic, helped provide the molecular basis for these findings. We think there will be further complexity to this regulation, creating the possibility for the discovery of additional mechanisms that regulate DNA repair pathways and response to therapy and potential new targets for diagnosis and therapy."
Journal information: Nature Structural & Molecular Biology
Provided by University of Pennsylvania School of Medicine