Forty's a crowd: Study shows that master regulator protein brings plethora of coactivators to gene expression sites

Molecular geneticists call big boss proteins that switch on broad developmental or metabolic programs "master regulators," as in master regulators of muscle development or fat metabolism. One such factor, the Activating Transcription Factor 6α (ATF6α) protein, takes charge following a cellular crisis known as endoplasmic reticulum (ER) stress, which is triggered by the accumulation of misfolded and aggregated proteins.
Molecularly, the ER stress pathway is always poised for action. Inactive ATF6α is normally embedded in cellular membranes, but at the first hint of protein overload, its working end springs superman-like into the nucleus, binds DNA and kicks on a host of target genes whose job is to clear a protein logjam.
Now, in a study published in the June 29 issue of The Journal of Biological Chemistry, and selected as "Paper of the Week" by the journal's editors, a team led by Stowers investigators Ron and Joan Conaway reveal that unlike the real superman ATF6α does not work solo. Using the ATF6α target gene HSPA5 as a probe, they apply mass spectrometry analysis to show that ATF6α recruits a fleet of coactivators to assist in target activation.
"We knew that as a master regulator, ATF6α was needed to turn on downstream genes in the ER stress response," says Ron Conaway, Ph.D., who with Joan Conaway, Ph.D., is co-corresponding author of the study. "Our goal was to determine what ATF6α was bringing with it to these genes' control elements."
"By devising a clever mix of state-of-the-art mass spectrometry and good old-fashioned biochemistry, this study has revealed that ATF6α is a virtual magnet for a wide range of 'A-list' co-regulators," said Michael K. Reddy, Ph.D., who oversees transcription mechanism grants at the National Institutes of Health's National Institute of General Medical Sciences, which partly supported the work. "These co-regulators offer a large array of proteins to target in efforts to control the ER stress response and to treat diseases that result from misfolded proteins."
That task of identifying co-regulators was challenging: labor-intensive molecular techniques the group applied to identify candidate interactors early on were not sensitive enough. At that point, the Conaways turned to their frequent collaborators Proteomics' Center director, Michael Washburn, Ph.D., and Laurence Florens, Ph.D., who heads the Stowers proteomics cores. Both had helped develop a sensitive mass spectrometry approach that can detect protein-protein interactions in highly complex mixtures, a technology known as MudPIT.
The group then set up a test-tube comparison. They genetically engineered a strand of DNA flanking the HSPA5 target gene, the so-called "enhancer" region recognized by ATF6α. They then dipped two identical DNA test strips into respective pots of cellular extracts—one containing ATF6α and one not—reasoning that factors in the ATF6α entourage would be recruited to the first but not the second. They then applied a single run of MudPIT to identify each ATF6α-specific partner.
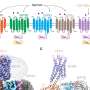
In short, they found that it takes not a village but a metropolis to activate an ATF6α target. Many proteins bound the enhancer in both samples, meaning either that they're just background, or else that they must bind DNA even when the gene is inactive. But more than 40 were present in about 5-fold excess only in ATF6αspiked samples, suggesting they are tethered to the enhancer by ATF6α.
Among the latter were components of a multi-subunit behemoth protein known as Mediator, which bridges specific genetic switches (like ATF6α) and the catalytic machinery that copies a gene. Other proteins recruited by ATF6α through overlapping but not identical domains belonged to other large complexes known as SAGA and ATAC, which enzymatically relax chromosome structure to allow gene expression.
Researchers know that all DNA-binding factors partner with other proteins to switch genes on or off. What is remarkable here is their sheer number. "It would be very interesting to find out whether this is the norm," says Ron Conaway. "This work raises a ton of little questions about mechanism."
Among them is how do ATF6α-interacting factors arrange themselves on the test strip, and does a single ATF6α bind to all of them at once? "There are three separate ATF6α binding sites on the HSPA5 enhancer and ATF6α itself forms a dimer," explains Dotan Sela, Ph.D., a Conaway lab postdoc and the study's first author, "So potentially within this region there could be as many six activation domains," he explains.
Solving these puzzles could reveal molecular targets for seemingly unrelated diseases. While a little ATF6α signaling is absolutely essential for cellular housekeeping, unrelieved ER stress is a hallmark of neurodegenerative conditions like Alzheimer's and Huntington's Diseases and is correlated with insulin insensitivity and type II diabetes.
A direct role for ATF6α in what some now call "misfolded protein diseases" is unclear. Nonetheless, the study suggests ways to dampen ER stress signaling molecularly. "We show that the Mediator is relevant to HSPA5 expression," says Sela. "So one way to keep ATF6α from turning on a gene might be to devise ways to block binding of the Mediator to ATF6α."
Joan Conaway also points out that MudPIT data analysis does not require previous identification of a "suspect." "Our approach complements methods that test candidate interactors one by one," says Joan Conaway. "Because the analysis is unbiased, it could reveal novel proteins interacting with a particular enhancer, which then could be confirmed using other methods."
The Conaways began their pioneering studies of mammalian gene expression over three decades ago, when only laborious biochemical techniques were available. As a result, both deeply appreciate what a technological leap the current work represents. "This study provides proof of principle for the utility of mass spectrometry in defining novel transcriptional activators," says Ron Conaway. "We want to compare this data with that from other activators—it's what we will be working on in the future."
Journal information: Journal of Biological Chemistry
Provided by Stowers Institute for Medical Research